- Review Article
- Open access
- Published:
Nonclassic Congenital Adrenal Hyperplasia
International Journal of Pediatric Endocrinology volume 2010, Article number: 625105 (2010)
Abstract
Nonclassic congenital adrenal hyperplasia (NCAH) due to P450c21 (21-hydroxylase deficiency) is a common autosomal recessive disorder. This disorder is due to mutations in the CYP21A2 gene which is located at chromosome 6p21. The clinical features predominantly reflect androgen excess rather than adrenal insufficiency leading to an ascertainment bias favoring diagnosis in females. Treatment goals include normal linear growth velocity and "on-time" puberty in affected children. For adolescent and adult women, treatment goals include regularization of menses, prevention of progression of hirsutism, and fertility. This paper will review key aspects regarding pathophysiology, diagnosis, and treatment of NCAH.
1. Introduction
Nonclassic congenital adrenal hyperplasia (NCAH) due to P450c21 (21-hydroxylase) deficiency is a common autosomal recessive disorder due to mutations in the CYP21A2 gene. This disorder was first described in 1957 by Decourt et al. [1]. Reported prevalences in women with androgen excess range from 0.6% to 9% (Table 1). Higher prevalences have been reported in Ashkenazi Jewish, Mediterranean, Middle-Eastern and Indian populations. Reported gene frequencies vary among ethnic groups and geographic region [2, 3].
NCAH due to mutations in other steroidogenic enzyme genes, such as 11 -hydroxylase (CYP11B1) and 3
-hydroxylase (CYP11B1) and 3 -hydroxysteroid dehydrogenase (HSD3B2), is extremely rare [4, 5]. The phenotypic spectrum for mutations in the cytochrome P450 oxidoreductase (POR) gene has been expanded to include amenorrhea, infertility, and low sex steroid hormone levels [6]. Partial loss of function missense mutations in the steroidogenic acute regulatory protein (StAR) gene has been associated with nonclassic lipoid adrenal hyperplasia; mutations in the ACTH receptor (MC2R) gene or the melanocortin 2 receptor accessory protein (MRAP) gene are associated with phenotypes similar to nonclassic lipoid adrenal hyperplasia [7]. This review will focus on NCAH due to CYP21A2 mutations.
-hydroxysteroid dehydrogenase (HSD3B2), is extremely rare [4, 5]. The phenotypic spectrum for mutations in the cytochrome P450 oxidoreductase (POR) gene has been expanded to include amenorrhea, infertility, and low sex steroid hormone levels [6]. Partial loss of function missense mutations in the steroidogenic acute regulatory protein (StAR) gene has been associated with nonclassic lipoid adrenal hyperplasia; mutations in the ACTH receptor (MC2R) gene or the melanocortin 2 receptor accessory protein (MRAP) gene are associated with phenotypes similar to nonclassic lipoid adrenal hyperplasia [7]. This review will focus on NCAH due to CYP21A2 mutations.
2. Molecular Genetics
To date, 127 mutations have been reported in CYP21A2 (http://www.hgmd.cf.ac.uk/); these mutations range from complete loss of enzyme function to partial enzyme activity. Most of the mutations result from recombination between the active gene, CYP21A2, and its highly homologous non-functional pseudogene, CYP21A1P(i.e., gene conversion), which is located in close proximity within the HLA region on chromosome 6p21.3. Nevertheless, approximately 10–12 mutations account for the majority of the affected alleles. The majority of the CYP21A2 mutations reported to date are associated with simple virilizing or salt-wasting classic congenital adrenal hyperplasia (CAH). Functional studies indicate that these mutations result in 0–5% residual enzymatic function [34].
Functional analysis of mutations associated with NCAH generally indicates a 50–80% loss of enzymatic (21-hydroxylase) function. Individuals with NCAH are generally compound heterozygotes bearing different CYP21A2 mutations on each allele. The missense mutation, V281L, accounts for at least one of the CYP21A2 alleles for most patients with NCAH. This genetic variant is commonly identified among Eastern Europeans especially those of Ashkenazi Jewish descent. Other missense mutations associated with NCAH include P30L, P453S, and R339H. Novel mutations associated with NCAH include R369W and I230T [35]. One-half to two-thirds of individuals with NCAH carry one allele encoding for a severe defect in enzyme function (which would result in classic CAH if present on both alleles) and an allele encoding a mild defect in enzyme function on the other allele. Roughly, phenotype correlates with molecular genotype and reflects the residual activity of the milder mutation [36–38]. Nevertheless, utilizing rigid criteria to distinguish among salt wasting, simple virilizing and NCAH can be problematic because impaired 21-hydroxylase function represents a continuum of decreased enzyme activity.
3. Pathophysiology
In broad terms, the virilizing forms (simple virilizing, salt-wasting, and nonclassic) of CAH are characterized by mutations that significantly impair cortisol biosynthesis and lead to the accumulation of steroid intermediates proximal to the deficient enzyme. The resulting loss of cortisol negative feedback inhibition leads to increased hypothalamic corticotrophin releasing hormone (CRH) and pituitary adrenocorticotrophic hormone (ACTH) secretion. With decreased P450c21 activity, conversions of 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol, and progesterone (P4) to deoxycorticosterone, are impaired. Elevated 17-OHP, P4, and androstenedione concentrations are typically found. The excessive ACTH stimulation also results in fasciculata-reticularis zone hypertrophy, resulting in the adrenal hyperplasia typical of the syndrome, and possibly increased adrenocortical nodularity. Individuals with NCAH generally have adequate mineralocorticoid secretion.
Unfortunately, the pathophysiology of NCAH (and CAH) is more complicated than this description would suggest. For example, patients with NCAH usually have no evidence of ACTH or CRH excess. In fact, some have an over-responsive glucocorticoid response to ACTH stimulation, possibly reflective of subtle adrenal hyperplasia [39]. Another mechanism resulting in excessive adrenal androgen secretion especially in NCAH results from the alteration in enzyme kinetics due to the CYP21A2 missense mutations [40]. The mutated enzyme protein is synthesized, but is less efficient than the wild type. The net result is an increased precursor to product ratio, independent of ACTH levels. Hence, P4 and 17-OHP levels in these patients may remain above normal even in the presence of excessive glucocorticoid administration [41]. In addition, genetic variations at other loci may influence steroid metabolism and steroid responsiveness.
Alterations in ovarian and gonadotropic function, with the appearance of a polycystic ovary-like phenotype, also contribute to the androgen excess of these patients [42, 43]. Functional ovarian abnormalities in patients with CAH and/or NCAH may relate to a number of etiologies, including disruption of the hypothalamic-pituitary-ovarian (HPO) axis by persistently elevated progesterones (e.g. P4 and/or 17-OHP) or androgens, and/or a direct glucocorticoid effect. Androgen excess impairs hypothalamic sensitivity to progesterone resulting in a persistently rapid GnRH pulse frequency which favors LH hypersecretion [44]. This LH hypersecretion initiates and maintains a vicious cycle in which excessive ovarian androgen secretion intensifies the consequences of the excessive adrenal androgen production. In fact, women with NCAH demonstrate higher LH concentrations than normal women [42]. Prenatal programming of the hypothalamus due to excessive in utero androgen exposure may contribute to LH hypersecretion and reproductive dysfunction among women with classical forms of CAH [45, 46]. However, in utero exposure to excessive androgens is unlikely to play a major role in the pathophysiology among women with NCAH.
Finally, while the 17,20-lyase activity of P450c17 towards  substrates (conversion of 17-OHP to androstenedione) is not significant in humans, it is possible that patients with CAH and NCAH may experience increased androgen excess due to a backdoor or alternative pathway converting either P4 or 17-OHP to more potent androgens such as dihydrotestosterone (DHT) [47]. Enzymes involved in this alternative pathway include 5
substrates (conversion of 17-OHP to androstenedione) is not significant in humans, it is possible that patients with CAH and NCAH may experience increased androgen excess due to a backdoor or alternative pathway converting either P4 or 17-OHP to more potent androgens such as dihydrotestosterone (DHT) [47]. Enzymes involved in this alternative pathway include 5 -reductases and 3
-reductases and 3 -hydroxysteroid dehydrogenases. The ovarian expression of 5
-hydroxysteroid dehydrogenases. The ovarian expression of 5 -reductase may contribute to excessive ovarian androgen secretion in NCAH as well as PCOS [48].
-reductase may contribute to excessive ovarian androgen secretion in NCAH as well as PCOS [48].
Overall, a more thorough understanding of the pathophysiologic mechanisms underlying the symptomatology of NCAH will improve our ability to select effective therapeutic regimens and choose reliable markers indicative of therapeutic success. For example, available data would suggest that the measurement of P4 or 17-OHP may not be the most accurate marker of therapeutic efficacy, and suppression of excessive androgen secretion from both ovaries and adrenals may be necessary for optimum steroidogenic control.
4. Clinical Features
Individuals with NCAH generally present with signs and symptoms of androgen excess rather than symptoms reflecting glucocorticoid deficiency. Children may present with premature pubarche (i.e. the development of pubic hair, axillary hair, and/or increased apocrine odor prior to age 8 years in girls and age 9 years in boys). In one study, 4.2% of 238 French children with premature pubarche were found to have NCAH; 17-OHP, androstenedione, and testosterone concentrations were significantly elevated among the children with NCAH compared to the remainder [49]. In a multicenter study including 220 individuals with NCAH, 92%, 8%, and 4% of patients diagnosed under the age of 10 years, 10–19 years, and 20–29 years, respectively, had a history of premature adrenarche [50].
Additional features in children include tall stature, accelerated linear growth velocity, and advanced skeletal maturation. Examination of the external genitalia may reveal clitoral enlargement in some girls without genital ambiguity. Phallic enlargement with prepubertal testes may be noted in boys. Although tall as children, the accelerated skeletal maturation promotes premature epiphyseal fusion leading to short stature in adulthood. Typically, these symptoms are more prominent among children with classic CAH.
During adolescence and adulthood, an ascertainment bias favors the diagnosis in females due to the nature of the hyperandrogenic symptoms. Symptoms include hirsutism, acne, alopecia, anovulation, and menstrual dysfunction. In a multicenter study, the most common symptoms among adolescent and adult women were hirsutism (59%), oligomenorrhea (54%), and acne (33%) [50]. Presenting symptoms in 161 women with NCAH were hirsutism (78%), menstrual dysfunction (54.7%), and decreased fertility (12%) [51].
Not all individuals with NCAH will be symptomatic. A study of the phenotype/genotype relationship in 330 family members revealed 9 symptomatic affected individuals, 42 clinically asymptomatic affected individuals, 242 heterozygotic carriers, and 37 unaffected individuals [51]. As found in this study, affected males are generally asymptomatic and usually identified following the diagnosis of a female family member. Peripubertal gynecomastia and adrenocortical incidentaloma are extremely uncommon presenting features [52, 53].
4.1. Acne
Acne can occur among patients with hyperandrogenism and may be the primary clinical manifestation of CAH. Severe cystic acne refractory to oral antibiotics and retinoic acid has been attributed to NCAH.
4.2. Alopecia
Additionally, male pattern baldness in young women with this disorder has been noted as the sole presenting symptom. Severe androgenic alopecia in association with marked virilization has also been reported in older women.
4.3. Hirsutism
Hirsutism is defined as the excessive growth of coarse terminal hairs in androgen-dependent areas. Hirsutism must be distinguished from hypertrichosis which is defined as generalized excessive growth of androgen-independent hair, and may be related to the use of certain medications (e.g., phenytoin, minoxidil, diazoxide, glucocorticoids, and cyclosporine), familial factors, or metabolic disorders (e.g., thyroid disturbances and anorexia nervosa).
The modified Ferriman-Gallwey score provides a semi-subjective method to assess the magnitude of hair growth in nine androgen-dependent areas such as the mustache area, chin, upper chest, abdomen, and back [54]. Although a modified Ferriman-Gallwey score of 6 to 8 is usually considered to indicate hirsutism, variation among ethnic groups occurs. Cosmetic treatments may reduce the ability to clinically detect hirsutism. Whereas hirsutism is uncommon in children or young adolescents, the prevalence of hirsutism and alopecia tends to increase over time [55]. Virilization and masculinization are terms used to describe the presence of more severe symptoms of androgen excess. Specifically, these terms refer to the presence of clitoromegaly, masculine body habitus, male pattern hair loss, and voice changes. These features, with the exception of occasional mild clitoromegaly, are not typically present in NCAH patients.
4.4. Ovulation, Menstruation and Reproductive Function
Women with NCAH often present with amenorrhea (primary or secondary), chronic anovulation, and infertility. Ultrasonography may demonstrate ovarian morphology reminiscent of polycystic ovary syndrome (PCOS). Polycystic ovary morphology may be present in about half of women with NCAH [56].
Many women with NCAH are relatively fertile [57, 58]. However, NCAH carries a greater risk of subfertility, in part due to the prevailing ovulatory dysfunction. Reports in women with classic CAH suggest that elevated progesterone concentrations play an important role in preventing menstrual cyclicity and fecundity [59, 60]. Likewise, persistently elevated levels of progesterones during the follicular phase in women with NCAH may interfere with the quality of cervical mucus, impeding penetration by sperm. In addition, elevated levels of 17-OHP and/or P4 during the preovulary (follicular) phase of the menstrual cycle may result in inadequate endometrial maturation and impaired embryo implantation.
Among 203 pregnancies in 101 women with NCAH, 138 pregnancies preceded the mother's diagnosis of NCAH. Spontaneous miscarriages were more common in the pregnancies prior to NCAH diagnosis [57]. Another series of women with NCAH desiring pregnancy reported similar findings with a decrease in spontaneous miscarriages during glucocorticoid treatment [61]. Potential limitations of these studies are that both are retrospective and largely include women ascertained by reproductive endocrinologists.
Since 21-hydroxylase deficient NCAH is an autosomal recessive disorder, the recurrence risk is 25% for pregnancies of the biological parents of the proband. Thus, siblings of the proband may benefit from diagnostic evaluation for NCAH. For women with NCAH, the risk of having a child with salt-losing or simple virilizing classical forms of CAH depends in part on the probability that the father is a carrier and mother's genotype. Moran et al. found that the prevalence of 21-OH-deficiency among liveborn children was 2.5% which was higher than the 0.2% calculated prevalence. In addition, at the time of the study 15% of children of mothers with NCAH had been also diagnosed with NCAH [57]. Bidet et al. also found the prevalence of CAH to be greater than anticipated [61]. The suggested explanation for the higher than expected prevalence of CAH and NCAH in these populations may be the tendency for affected individuals to marry within their own ethnic background; some ethnic groups are enriched for CYP21A2 variants.
5. Other Considerations
5.1. Precocious Puberty
Although more commonly observed in children with classic CAH, skeletal maturation may be significantly advanced among children with NCAH and may be associated with gonadotropin-dependent precocious puberty [62]. Typically, the signs and symptoms of puberty become conspicuous after the initiation of glucocorticoid treatment. In this situation, the hypothalamic GnRH pulse generator prematurely resumes pulsatile GnRH secretion leading to increased LH and FSH secretion resulting in increased gonadal steroid production. The precocious puberty is considered to be secondary to the excessive adrenal steroid secretion and advanced skeletal maturation associated with NCAH. Some children with secondary GnRH-dependent precocious puberty benefit from treatment with GnRH-super agonists such as leuprolide acetate or histrelin.
5.2. Bone Mineral Density
Glucocorticoids influence bone metabolism by suppressing osteoblast activity, promoting increased bone resorption by osteoclasts, and interfering with calcium absorption from the gastrointestinal tract [63]. Thus, the need for chronic glucocorticoid therapy leads to concerns regarding bone density for individuals with CAH. Since DXA is based on a two-dimensional technique, interpretation of areal bone mineral density assessed by DXA scan can be confounded by bone width and height. Thus, DXA can underestimate bone mineral density in shorter individuals [64]. Available data, derived from outcome reports for individuals with classic CAH, are inconsistent due to varying glucocorticoid doses, potential compliance issues, and subject heterogeneity [65, 66]. In theory, inadequate treatment would lead to androgen excess that would be anticipated, in turn, to increase BMD. On the other hand, excessive glucocorticoid replacement treatment would be expected decrease BMD. At this time, outcome data regarding BMD in NCAH are limited. Nevertheless, it has been suggested that maintaining vitamin D sufficiency should be a goal for individuals with CAH [67].
5.3. Gonadal Rest Tumors
During early gestation, cells destined to become the steroid producing cells of the adrenal cortex and gonads differentiate from neighboring regions of the coelomic epithelium. Subsequently, some adrenal precursor cells migrate, descend into the scrotum with the testes, retain ACTH responsiveness, and can develop into testicular adrenal rest tumors (TARTs) [68]. Such tumors have generally been described in boys or men with classic CAH and poor compliance [69]. Incomplete detection and underdiagnosis of NCAH in men hinders accurate ascertainment of the frequency of TARTs in men with NCAH.
5.4. Adrenal Tumors
Adrenal tumors have rarely been identified among individuals with NCAH. Following discovery of an adrenal incidentaloma, an 88 year old women was diagnosed with NCAH; her genetic analysis showed V281L and I172N [70]. A 57 year old man ascertained by finding an adrenal incidentaloma was diagnosed with NCAH; he had elevated serum 17-OHP concentrations and urinary 17-ketosteroid excretion [71]. Adrenal myelolipomas have been reported among untreated adults with NCAH [72].
5.5. Contiguous Gene Deletion Syndrome
The CYP21A2 and CYP21A1P genes map to the HLA complex at chromosome 6p21. Another gene located in this region of the genome encodes for tenascin-X (TNXB). Tenascin-X is a large extracellular matrix protein which is expressed in the dermis of the skin, and cardiac and skeletal connective tissue. Loss of function TNXB mutations are associated with hypermobility Ehlers-Danlos syndrome [73]. Individuals with CYP21A2 deletions may have haploinsufficiency for TNXB and may manifest joint hypermobility, joint subluxations, and chronic musculoskeletal pain [74]. A girl with classic CAH was found to have a quadricuspid aortic valve, single kidney, bicornuate uterus, and vesicoureteral reflux [75]. Thus, it is possible that the Ehlers-Danlos phenotype could occur among patients with NCAH who carry a continuous gene deletion involving this region on one allele and V281L on the other allele. Nevertheless, to date, no such case has been reported.
5.6. Metabolic Consequences
Factors associated with increased risk for metabolic consequences cluster in women with NCAH. These factors include obesity, hypertension, and insulin resistance. The androgen excess may independently contribute to this risk due to atherogenic lipid profiles. Using the minimal model to assess insulin sensitivity, insulin sensitivity was found to be decreased in six untreated nonobese women with NCAH compared to control subjects [76]. Comparison of metabolic parameters in women with PCOS, women with NCAH, and healthy control women showed that metabolic parameters were comparable among women with NCAH, lean women with PCOS, and healthy control women whereas metabolic dysfunction was evident in the obese women with PCOS [20]. Available studies regarding insulin sensitivity have provided inconsistent results and generally involve women with classical forms of CAH [77, 78].
Platelet dysfunction is another feature that can be associated with insulin resistance. To distinguish between the consequence of hyperandrogenism and hyperinsulinism, agonist-induced platelet function was studied. Whereas platelet aggregation in samples from women with PCOS was high, platelet aggregation in samples from women with NCAH was comparable to the healthy controls [79].
6. Psychosocial Considerations and Quality of Life
Gender role develops as a result of society's expectations concerning behavior. Prenatal factors such as hormones and environmental exposures are hypothesized to influence gender role. Yet, the specific details regarding how prenatal androgen exposure affects gender identity of girls with classic CAH remain to be clarified [80, 81]. Most women with adrenal hyperplasia, both CAH and NCAH, demonstrate heterosexual preferences [82]. Despite the impression that women with NCAH primarily have postnatal androgen excess, the frequency of homosexuality and bisexuality was slightly increased in one study compared to non-CAH controls [82]. Limitations of this study include small sample size and cross-sectional design. It is also unclear how representative the subjects are relative to other women with NCAH.
Infertility is inextricably related to self-esteem and psychosocial adjustment. The anatomic concerns related to women with classic CAH such as pain with vaginal penetration are generally not germane for women with NCAH [83]. As surgical, medical, and psychological treatments have improved, more women with classic and NCAH have successfully completed pregnancies and given birth [84].
7. Diagnosis
Individuals with the salt-wasting and simple virilizing forms of CAH are generally recognized in the newborn period, and most affected females are detected by the genital ambiguity. Without a family history, males with classic CAH are identified through newborn screening programs. In general, newborn screening programs fail to detect individuals with NCAH [85].
Newborn screening programs measure of 17-OHP in whole blood spots collected on filter paper. The 17-OHP results for infants with NCAH are often not as elevated as those for infants with classic CAH. Another confounder is that preterm infants, heterozygotic carriers, and sick infants have 17-OHP concentrations which overlap the concentrations measured in infants with NCAH. Thus, imperfect recall occurs almost by design because of the need to avoid excessive numbers of false positive results while trying to maintain adequate sensitivity and specificity to detect infants with classic CAH. Given the comparatively mild course of NCAH during childhood and the anxiety and costs involved in false positive results, treatment based solely on elevated hormone levels in the absence of symptoms may only increase the risk for iatrogenic adrenal insufficiency without any clear therapeutic benefit. Thus, risk/benefit analysis of confirming the diagnosis of NCAH in a neonate prior to the development of symptoms is unresolved due to the lack of outcome data [86].
The clinical features of NCAH in postpubertal adults may be difficult to differentiate from those of the polycystic ovary syndrome (PCOS) or, in children, from premature adrenarche. Although random 17-OHP concentrations are usually diagnostic in classical forms of CAH, random 17-OHP concentrations may be within the normal range for individuals with NCAH. Thus, the acute ACTH stimulation test remains the gold standard to confirm decreased 21-hydroxylase activity. Following collection of a blood sample to measure baseline hormone concentrations, synthetic ACTH (Cortrosyn, 0.25 mg) is administered. A second blood sample is collected 30–60 minutes later. Correlation of hormone concentrations with genetic analyses has suggested that mutations are likely to be identified on both alleles when the ACTH-stimulated 17-OHP value exceeds 1500 ng/dL, although a few NCAH patients, particularly if older, will demonstrate ACTH-stimulated 17-OHP levels between 1000 and 1500 ng/dL. In one study, among 123 women with NCAH confirmed by molecular CYP21A2 analysis, mean basal 17-OHP and mean ACTH-stimulated 17-OHP concentrations were 1300  1420 ng/dL and 4080
1420 ng/dL and 4080  2040 ng/dL, respectively [51].
2040 ng/dL, respectively [51].
In general, it is impractical to perform an acute ACTH stimulation test in all women suspected of NCAH (e.g. those with hyperandrogenic features or ovulatory/menstrual dysfunction). Various investigators have suggested the use of unstimulated levels of 17-OHP as a predictor of NCAH [24, 57, 87, 88]. Levels of 170–300 ng/dL have been found to be useful as a screening tool, best if obtained in the morning and, most importantly (to reduce false positives), in the follicular (preovulatory) phase of the menstrual cycle Among 129 Portuguese women with hyperandrogenism and menstrual dysfunction, 87% of women with NCAH, 25% of lean women with PCOS, 20% of obese women with PCOS, and 7% of control women had basal 17-OHP concentrations  200 ng/dL [20]. In childhood, NCAH may present with premature adrenarche. In a sample of 238 French children with premature pubic hair, of which 4.2% had NCAH, the use of a 17-OHP cut-off value of greater than 200 ng/dL provided a 100% sensitivity and 99% sensitivity for the detection of NCAH in this cohort [49].
200 ng/dL [20]. In childhood, NCAH may present with premature adrenarche. In a sample of 238 French children with premature pubic hair, of which 4.2% had NCAH, the use of a 17-OHP cut-off value of greater than 200 ng/dL provided a 100% sensitivity and 99% sensitivity for the detection of NCAH in this cohort [49].
Genetic testing should not be considered a first-line diagnostic study in individuals suspected of NCAH. However, genetic studies may be useful in those patients (men or women) who are considering future fertility. Genetic testing can identify those individuals who are compound heterozygotes and may carry an allele encoding a severe defect in CYP21A2. Overall, screening using morning 17-OHP concentrations, obtained in the follicular phase in reproductive-aged females, and followed, if positive, by an acute ACTH stimulation remain the essential clinical tools to diagnose NCAH.
8. Laboratory Analysis
Laboratory techniques used to measure 17-OHP include radioimmunoassays (RIA), enzyme-linked immunosorbent assays (EIA) and time-resolved fluoroimmunoassays (FIA). While there may be variability between the laboratories, our studies indicate a high degree of correlation between laboratories [50]. As noted above, the presence of cross-reacting steroids of fetal adrenal origin may hinder the interpretation of 17-OHP concentrations in preterm and term infants. Recent technical improvement involve tandem mass spectrometry (MS) linked to liquid chromatography (LC) [89]. Detection of 17-OHP from dried whole blood spots using LC followed by tandem mass spectrometry (LC/MS/MS) has been reported [90]. The use of LC/MS/MS may improve the sensitivity and specificity of newborn screening. The use of LC/MS/MS is not only limited to newborns, the sensitivity and specificity of this technique make it applicable for 17-OHP determinations for children, adolescents, and adults.
When considering genetic testing, an important limitation is that molecular genetic analysis can be confounded by the complexity of the CYP21A2-CYP21A1P loci. Multiple mutations can occur on one allele so that the identification of two mutations does not always signify CAH because both mutations may occur on the same allele (cis). Copy number variation involving the CYP21A2-CYP21A1P region may result in multiple copies of CYP21A2 on a single allele. In addition, most commercially available screening panels assay for the 10–12 most common mutations, and may not be able to detect all mutations [91]. Inclusion of a DNA sample from at least one parent and/or a child may discriminate between variants on the same (cis) or different (trans) alleles.
9. Treatment
Treatment needs to be directed towards the symptoms. In other words, treatment should not be initiated merely to decrease abnormally elevated hormone concentrations. Clinical goals of treatment include normal linear growth velocity, normal rate of skeletal maturation, "on-time" puberty, and appropriate weight status for children and adolescents. For adolescent and adult women, goals of therapy include regularization of menstrual cycles, prevention of progressive hirsutism and acne, and fertility. For each child, adolescent, and adult with NCAH, the benefits of treatment should be weighed against the potential risks of acute adrenal insufficiency secondary to iatrogenic adrenal suppression due to glucocorticoid treatment. Treatment of hirsutism may also necessitate adjunctive cosmetic methods such as laser, electrolysis, and depilatories.
Glucocorticoid treatment can be utilized for children and adolescents with significantly advanced skeletal maturation. Although treatment with oral contraceptives alone may be sufficient in oligomenorrheic, acneic, or mildly hirsute adolescents and adult women not seeking fertility, early glucocorticoid treatment may be beneficial to decrease the risk of persistent anovulation. In a crossover design involving eight women with NCAH, oral contraceptive therapy was associated with increased SHBG and decreased free testosterone concentrations. As would be anticipated, menstrual cyclicity was restored with oral contraceptive therapy [92].
The use of anti-androgens (e.g. flutamide, cyproterone acetate, or finasteride) should also be considered in women complaining of excess unwanted hair growth or scalp hair loss (androgenic alopecia). Despite minimal changes in testosterone and androstenedione concentrations, greater improvement in hirsutism was noted with the use of cyproterone acetate compared to hydrocortisone among women with NCAH [93]. Thus, for adolescent and adult women with NCAH who are taking glucocorticoids, the addition of oral contraceptives or anti-androgens may allow for lower glucocorticoid dosage.
Among adults, treatment is generally reserved for symptomatic individuals, as the risks of iatrogenic adrenal suppression with glucocorticoid treatment need to be balanced against the potential benefits of treatment. Most untreated individuals with NCAH manifest an adequate response to stress. One effective regimen involves hydrocortisone, 6–15 mg/m2/day divided into three daily doses. Many clinicians suggest that reverse circadian dosing with the highest hydrocortisone dose at night provides improved control, but no consensus exists regarding how to divide the doses [56, 94]. All individuals on glucocorticoid therapy require instruction regarding stress doses, including parenteral therapy, and should wear medical alert identifying badges/jewelry.
For daily treatment, prednisone, prednisolone, or dexamethasone may also be used in adults. Because of the potential detrimental effect of glucocorticoids on the fetus [95], various practitioners suggest that a glucocorticoid that is inactivated by placental 11 -hydroxysteroid dehydrogenase type II (e.g. hydrocortisone, prednisone, and prednisolone) should be used in sexually active females who are not on a highly effective contraceptive [95] or who become pregnant, unless specifically intending to suppress the fetal adrenal (see below). As the androgen secretory potential of the adrenal cortex declines with age [96], the demand for glucocorticoids to suppress adrenal androgen secretion in NCAH may ameliorate with age. Stress doses of hydrocortisone are essential for affected individuals maintained on glucocorticoid treatment. For emergency situations, including labor and delivery and surgery, the empiric stress dose is parenteral hydrocortisone (Solu-Cortef), 100 mg, IV or IM.
-hydroxysteroid dehydrogenase type II (e.g. hydrocortisone, prednisone, and prednisolone) should be used in sexually active females who are not on a highly effective contraceptive [95] or who become pregnant, unless specifically intending to suppress the fetal adrenal (see below). As the androgen secretory potential of the adrenal cortex declines with age [96], the demand for glucocorticoids to suppress adrenal androgen secretion in NCAH may ameliorate with age. Stress doses of hydrocortisone are essential for affected individuals maintained on glucocorticoid treatment. For emergency situations, including labor and delivery and surgery, the empiric stress dose is parenteral hydrocortisone (Solu-Cortef), 100 mg, IV or IM.
Laboratory goals include normalization of androstenedione and testosterone levels; alternatively, suppression of DHEAS levels occurs with minimal doses of glucocorticoids. Normalization of 17-OHP or P4 concentrations indicates excessive hormone replacement therapy, unless so intended in patients seeking fertility (see below).
Women seeking fertility may benefit from the use of ovulatory agents (clomiphene or menotropins). Preliminarily, the preconception use of concomitant glucocorticoids appears to reduce the risk of miscarriages in NCAH patients conceiving [57]. In addition, in this setting maximum suppression of 17-OHP and P4 could potentially allow maximum endometrial proliferation and improve implantation. In addition, all women with NCAH desiring to conceive should undergo genetic screening to determine the presence of severe CYP21A2 mutations (and, hence, the risk of having a child with CAH), and if a severe allele is present then genetic screening of the father should also be undertaken. Many of these patients benefit from preconception genetic counseling.
In patients who conceive and whose child is considered at high risk for intrauterine virilization (i.e. a female infant) consideration may be given to using high dose dexamethasone suppression in early gestation, before 10 weeks gestational age [97, 98], until definitive diagnosis and chromosomal sex can be obtained using chorionic villi sampling or early amniocentesis (by 10–13 weeks gestational age). However, this experimental therapy is associated with significant risk of Cushingoid features and glucose intolerance in the mother [99]. The effects on the fetus particularly related to brain development are unknown. In addition, since all "at-risk" fetuses are treated until the definitive diagnosis and chromosomal sex are known, 7/8 fetuses are needlessly exposed to prenatal dexamethasone [100]. Overall, the risk of a patient with NCAH of having a child with CAH is relatively low (2.5%) [57]. Therefore, couples who conceive and whose genetic diagnosis is not known should not be considered for prenatal dexamethasone suppression, unless they have had a prior child with salt-losing or simple virilizing CAH. Those choosing to use prenatal dexamethasone should do so only as participants of IRB-approved research studies [101].
10. Conclusions
Nonclassic congenital adrenal hyperplasia is a common autosomal recessive disorder that can present in childhood, adolescence, and adulthood. The typical symptoms of hirsutism, oligomenorrhea, infertility, acne, and premature pubarche lead to an ascertainment bias in favor of identifying affected women. The nature of the symptoms leads to consideration of polycystic ovary syndrome in the differential diagnosis. Although NCAH is a genetic disorder, the use of morning follicular phase 17-OHP concentrations and acute ACTH stimulation tests are essential diagnostic studies due to the complexity of the CYP21A2 locus. Once the diagnosis is confirmed, genetic analysis may be useful. The specific treatment should be individualized and directed towards the individual's symptoms and current medical needs.
References
Decourt J, Jayle MF, Baulieu E: Virilisme cliniquement tardif avec excrétion de prégnanetriol et insuffisance de la production du cortisol. Annales d'Endocrinologie. 1957, 18 (3): 416-422.
Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A: High frequency of nonclassical steroid 21-hydroxylase deficiency. American Journal of Human Genetics. 1985, 37 (4): 650-667.
Wilson RC, Nimkarn S, Dumic M: Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Molecular Genetics and Metabolism. 2007, 90 (4): 414-421. 10.1016/j.ymgme.2006.12.005.
Lutfallah C, Wang W, Mason JI: Newly proposed hormonal criteria via genotypic proof for type II 3
 -hydroxysteroid dehydrogenase deficiency. Journal of Clinical Endocrinology and Metabolism. 2002, 87 (6): 2611-2622. 10.1210/jc.87.6.2611.
-hydroxysteroid dehydrogenase deficiency. Journal of Clinical Endocrinology and Metabolism. 2002, 87 (6): 2611-2622. 10.1210/jc.87.6.2611.Joehrer K, Geley S, Strasser-Wozak EMC: CYP11B1 mutations causing non-classic adrenal hyperplasia due to 11
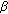 - hydroxylase deficiency. Human Molecular Genetics. 1997, 6 (11): 1829-1834. 10.1093/hmg/6.11.1829.
- hydroxylase deficiency. Human Molecular Genetics. 1997, 6 (11): 1829-1834. 10.1093/hmg/6.11.1829.Sahakitrungruang T, Huang N, Tee MK: Clinical, genetic, and enzymatic characterization of P450 oxidoreductase deficiency in four patients. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (12): 4992-5000. 10.1210/jc.2009-1460.
Metherell LA, Naville D, Halaby G: Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. Journal of Clinical Endocrinology and Metabolism. 2009, 94: 3865-3871. 10.1210/jc.2009-0467.
Emans SJ, Grace E, Fleischnick E, Mansfield MJ, Crigler JF: Detection of late-onset 21-hydroxylase deficiency congenital adrenal hyperplasia in adolescents. Pediatrics. 1983, 72 (5): 690-695.
Cobin RH, Futterweit W, Fiedler RP, Thornton JC: Adrenocorticotropic hormone testing in idiopathic hirsutism and polycystic ovarian disease: a test of limited usefulness. Fertility and Sterility. 1985, 44 (2): 224-226.
Azziz R, Zacur HA: 21-hydroxylase deficiency in female hyperandrogenismml: screening and diagnosis. Journal of Clinical Endocrinology and Metabolism. 1989, 69 (3): 577-584. 10.1210/jcem-69-3-577.
Azziz R, Bradley EL, Potter HD, Boots LR: 3
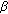 -Hydroxysteroid dehydrogenase deficiency in hyperandrogenism. American Journal of Obstetrics and Gynecology. 1993, 168 (3): 889-895.
-Hydroxysteroid dehydrogenase deficiency in hyperandrogenism. American Journal of Obstetrics and Gynecology. 1993, 168 (3): 889-895.Chetkowski RJ, DeFazio J, Shamonki I, Judd HL, Chang RJ: The incidence of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency among hirsute women. Journal of Clinical Endocrinology and Metabolism. 1984, 58 (4): 595-598. 10.1210/jcem-58-4-595.
Azziz R, Sanchez LA, Knochenhauer ES: Androgen excess in women: wxperience with over 1000 consecutive patients. Journal of Clinical Endocrinology and Metabolism. 2004, 89 (2): 453-462. 10.1210/jc.2003-031122.
Innanen VT, Vale JM: Assessment of the one hour adrenocorticotrophic hormone test in the diagnosis of attenuated 21-hydroxylase deficiency. Journal of Clinical Pathology. 1990, 43 (6): 493-495. 10.1136/jcp.43.6.493.
Romaguera J, Moran C, Diaz-Montes TP, Hines GA, Cruz RI, Azziz R: Prevalence of 21-hydroxylase-deficient nonclassic adrenal hyperplasia and insulin resistance among hirsute women from Puerto Rico. Fertility and Sterility. 2000, 74 (1): 59-62. 10.1016/S0015-0282(00)00566-5.
McLaughlin B, Barrett P, Finch T, Devlin JG: Late onset adrenal hyperplasia in a group of Irish females who presented with hirsutism, irregular menses and/or cystic acne. Clinical Endocrinology. 1990, 32 (1): 57-64. 10.1111/j.1365-2265.1990.tb03750.x.
Turner EI, Watson MJ, Perry LA, White MC: Investigation of adrenal function in women with oligomenorrhoea and hirsutism (clinical PCOS) from the north-east of England using an adrenal stimulation test. Clinical Endocrinology. 1992, 36 (4): 389-397. 10.1111/j.1365-2265.1992.tb01465.x.
Kuttenn F, Couillin P, Girard F: Late-onset adrenal hyperplasia in hirsutism. The New England Journal of Medicine. 1985, 313 (4): 224-231. 10.1056/NEJM198507253130404.
Blanché H: Exhaustive screening of the 21-hydroxylase gene in a population of hyperandrogenic women. Human Genetics. 1997, 101: 56-60. 10.1007/s004390050586.
Pall M, Azziz R, Beires J, Pignatelli D: The phenotype of hirsute women: a comparison of polycystic ovary syndrome and 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertility and Sterility. In press
Carmina E, Malizia G, Janni A: Prevalence of late-onset 21-hydroxylase deficiency in hirsutism of Western Sicily. Journal of Endocrinological Investigation. 1987, 10 (supplement, article 75):
Motta P, Catania A, Airaghi L, Mangone I, Cantalamessa L, Zanussi C: Prevalence of late-onset adrenal hyperplasia in postmenarchal hirsutism. Journal of Endocrinological Investigation. 1988, 11 (9): 675-678.
Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA: Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. Journal of Clinical Endocrinology and Metabolism. 2006, 91: 2-6. 10.1210/jc.2005-1457.
Escobar-Morreale HF, Sanchón R, San Millán JL: A prospective study of the prevalence of nonclassical congenital adrenal hyperplasia among women presenting with hyperandrogenic symptoms and signs. Journal of Clinical Endocrinology and Metabolism. 2008, 93 (2): 527-533. 10.1210/jc.2007-2053.
Fanta M, Cibula D, Vrbíková J: Prevalence of nonclassic adrenal hyperplasia (NCAH) in hyperandrogenic women. Gynecological Endocrinology. 2008, 24 (3): 154-157. 10.1080/09513590801911992.
Trakakis E, Rizos D, Loghis C: The prevalence of non-classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency in greek women with hirsutism and polycystic ovary syndrome. Endocrine Journal. 2008, 55 (1): 33-39. 10.1507/endocrj.K07-053.
Akinci A, Yordam N, Ersoy F, Uluşahin N, Oğuz H: The incidence of non-classical 21 -hydroxylase deficiency in hirsute adolescent girls. Gynecological Endocrinology. 1992, 6 (2): 99-106. 10.3109/09513599209046392.
Yarman S, Dursun A, Oğuz F, Alagol F: The prevalence, molecular analysis and HLA typing of late-onset 21-hydroxylase deficiency in Turkish woman with hirsutism and polycystic ovary. Endocrine Journal. 2004, 51 (1): 31-36. 10.1507/endocrj.51.31.
Unluhizarci K, Kula M, Dundar M: The prevalence of non-classic adrenal hyperplasia among Turkish women with hyperandrogenism. Gynecological Endocrinology. 2010, 26 (2): 139-143. 10.3109/09513590903215466.
Kamel N, Tonyukuk V, Emral R, Corapçioğlu D, Baştemir M, Güllü S: The prevalence of late onset congenital adrenal hyperplasia in hirsute women from central anatolia. Endocrine Journal. 2003, 50 (6): 815-823. 10.1507/endocrj.50.815.
Eldar-Geva T, Hurwitz A, Vecsei P, Palti Z, Milwidsky A, Rösler A: Secondary biosynthetic defects in women with late-onset congenital adrenal hyperplasia. The New England Journal of Medicine. 1990, 323 (13): 855-863. 10.1056/NEJM199009273231302.
Mithal A, Ammini AC, Godbole MM: Late-onset adrenal hyperplasia in North Indian hirsute women. Hormone Research. 1988, 30 (1): 1-4. 10.1159/000181016.
Khandekar S, Lata V, Dash RJ: Screening for late onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Indian Journal of Medical Research. Section B. 1990, 92: 79-82.
Miller WL: Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1994, 78 (2): 241-246. 10.1210/jc.78.2.241.
Tardy V, Menassa R, Sulmont V: Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patients affected with 21-hydroxylase deficiency and in one carrier. Journal of Clinical Endocrinology and Metabolism. 2010, 95 (3): 1288-1300. 10.1210/jc.2009-1202.
Speiser PW, Dupont J, Zhu D: Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Investigation. 1992, 90 (2): 584-595. 10.1172/JCI115897.
Wedell A, Ritzén EM, Haglund-Stengler B, Luthman H: Steroid 21-hydroxylase deficiency: three additional mutated alleles and establishment of phenotype-genotype relationships of common mutations. Proceedings of the National Academy of Sciences of the United States of America. 1992, 89 (15): 7232-7236. 10.1073/pnas.89.15.7232.
Jääskeläinen J, Levo A, Voutilainen R, Partanen J: Population-wide evaluation of disease manifestation in relation to molecular genotype in steroid 21-hydroxylase (CYP21) deficiency: good correlation in a well defined population. Journal of Clinical Endocrinology and Metabolism. 1997, 82 (10): 3293-3297. 10.1210/jc.82.10.3293.
Huerta R, Dewailly D, Decanter C, Knochenhauer ES, Boots LR, Azziz R: Adrenocortical hyperresponsivity to adrenocorticotropic hormone: a mechanism favoring the normal production of cortisol in 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertility and Sterility. 2000, 74 (2): 329-334. 10.1016/S0015-0282(00)00631-2.
Azziz R, Slayden SM: The 21-hydroxylase-deficient adrenal hyperplasias: more than ACTH oversecretion. Journal of the Society for Gynecologic Investigation. 1996, 3 (6): 297-302. 10.1016/S1071-5576(96)00044-5.
Sánchez LA, Moran C, Reyna R, Ochoa T, Boots LR, Azziz R: Adrenal progestogen and androgen production in 21-hydroxylase-deficient nonclassic adrenal hyperplasia is partially independent of adrenocorticotropic hormone stimulation. Fertility and Sterility. 2002, 77 (4): 750-753. 10.1016/S0015-0282(01)03236-8.
Levin JH, Carmina E, Lobo RA: Is the inappropriate gonadotropin secretion of patients with polycystic ovary syndrome similar to that of patients with adult-onset congenital adrenal hyperplasia?. Fertility and Sterility. 1991, 56 (4): 635-640.
Carmina E, Lobo RA: Ovarian suppression reduces clinical and endocrine expression of late- onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertility and Sterility. 1994, 62 (4): 738-743.
Blank SK, McCartney CR, Helm KD, Marshall JC: Neuroendocrine effects of androgens in adult polycystic ovary syndrome and female puberty. Seminars in Reproductive Medicine. 2007, 25 (5): 352-359. 10.1055/s-2007-984741.
Barnes RB, Rosenfield RL, Ehrmann DA: Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. Journal of Clinical Endocrinology and Metabolism. 1994, 79 (5): 1328-1333. 10.1210/jc.79.5.1328.
Abbott DH, Barnett DK, Bruns CM, Dumesic DA: Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome?. Human Reproduction Update. 2005, 11 (4): 357-374. 10.1093/humupd/dmi013.
Ghayee HK, Auchus RJ: Basic concepts and recent developments in human steroid hormone biosynthesis. Reviews in Endocrine and Metabolic Disorders. 2007, 8 (4): 289-300. 10.1007/s11154-007-9052-2.
Auchus RJ: Non-traditional metabolic pathways of adrenal steroids. Reviews in Endocrine and Metabolic Disorders. 2009, 10 (1): 27-32. 10.1007/s11154-008-9095-z.
Armengaud JB, Charkaluk ML, Trivin C: Precocious pubarche: distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. Journal of Clinical Endocrinology and Metabolism. 2009, 94: 2835-2840. 10.1210/jc.2009-0314.
Moran C, Azziz R, Carmina E: 21-hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. American Journal of Obstetrics and Gynecology. 2000, 183 (6): 1468-1474. 10.1067/mob.2000.108020.
Bidet M, Bellanné-Chantelot C, Galand-Portier M-B: Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (5): 1570-1578. 10.1210/jc.2008-1582.
Wasniewska M, Raiola G, Galati MC: Non-classical 21-hydroxylase deficiency in boys with prepubertal or pubertal gynecomastia. European Journal of Pediatrics. 2008, 167 (9): 1083-1084. 10.1007/s00431-007-0625-6.
Nigawara T, Kageyama K, Sakihara S: A male case of nonclassical 21-hydroxylase deficiency first manifested in his sixties with adrenocortical incidentaloma. Endocrine Journal. 2008, 55 (2): 291-297. 10.1507/endocrj.K07-119.
Hatch R, Rosenfield RL, Kim MH, Tredway D: Hirsutism: implications, etiology, and management. American Journal of Obstetrics and Gynecology. 1981, 140 (7): 815-830.
Moran C, Azziz R, Carmina E: 21-hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. American Journal of Obstetrics and Gynecology. 2000, 183 (6): 1468-1474. 10.1067/mob.2000.108020.
Azziz R, Dewailly D, Owerbach D: Nonclassic adrenal hyperplasia: current concepts. Journal of Clinical Endocrinology and Metabolism. 1994, 78 (4): 810-815. 10.1210/jc.78.4.810.
Moran C, Azziz R, Weintrob N: Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2006, 91 (9): 3451-3456. 10.1210/jc.2006-0062.
Feldman S, Billaud L, Thalabard J-C: Fertility in women with late-onset adrenal hyperplasia due to 21- hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1992, 74 (3): 635-639. 10.1210/jc.74.3.635.
Stikkelbroeck NMML, Sweep CGJ, Braat DDM, Hermus ARMM, Otten BJ: Monitoring of menstrual cycles, ovulation, and adrenal suppression by saliva sampling in female patients with 21-hydroxylase deficiency. Fertility and Sterility. 2003, 80 (4): 1030-1036. 10.1016/S0015-0282(03)01006-9.
Casteràs A, De Silva P, Rumsby G, Conway GS: Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clinical Endocrinology. 2009, 70: 833-837. 10.1111/j.1365-2265.2009.03563.x.
Bidet M, Bellanné-Chantelot C, Galand-Portier M-B: Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2010, 95 (3): 1182-1190. 10.1210/jc.2009-1383.
Pescovitz OH, Comite F, Cassorla F: True precocious puberty complicating congenital adrenal hyperplasia: treatment with a luteinizing hormone-releasing hormone analog. Journal of Clinical Endocrinology and Metabolism. 1984, 58 (5): 857-861. 10.1210/jcem-58-5-857.
Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A: Glucocorticoid-induced osteoporosis: an update. Trends in Endocrinology and Metabolism. 2006, 17 (4): 144-149. 10.1016/j.tem.2006.03.009.
Leonard MB: Glucocorticoid-induced osteoporosis in children: impact of the underlying disease. Pediatrics. 2007, 119 (supplement 2): S166-S174. 10.1542/peds.2006-2023J.
Chakhtoura Z, Bachelot A, Samara-Boustani D: Impact of total cumulative glucocorticoid dose on bone mineral density in patients with 21-hydroxylase deficiency. European Journal of Endocrinology. 2008, 158 (6): 879-887. 10.1530/EJE-07-0887.
Falhammar H, Filipsson H, Holmdahl G: Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (12): 4643-4649. 10.1210/jc.2007-0744.
Bachelot A, Chakthoura Z, Rouxel A, Dulon J, Touraine P: Classical forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency in adults. Hormone Research. 2008, 69 (4): 203-211. 10.1159/000113020.
Sullivan JG, Gohel M, Kinder RB: Ectopic adrenocortical tissue found at groin exploration in children: incidence in relation to diagnosis, age and sex. BJU International. 2005, 95 (3): 407-410. 10.1111/j.1464-410X.2005.05310.x.
Claahsen-van der Grinten HL, Otten BJ, Stikkelbroeck MML, Sweep FCGJ, Hermus ARMM: Testicular adrenal rest tumours in congenital adrenal hyperplasia. Best Practice and Research. 2009, 23 (2): 209-220. 10.1016/j.bpg.2009.03.006.
Falhammar H, Thorén M: An 88-year-old woman diagnosed with adrenal tumor and congenital adrenal hyperplasia: connection or coincidence?. Journal of Endocrinological Investigation. 2005, 28 (5): 449-453.
Nagasaka S, Kubota K, Motegi T: A case of silent 21-hydroxylase deficiency with persistent adrenal insufficiency after removal of an adrenal incidentaloma. Clinical Endocrinology. 1996, 44 (1): 111-116. 10.1046/j.1365-2265.1996.631456.x.
Murakami C, Ishibashi M, Kondo M: Adrenal myelolipoma associated with congenital adrenal 21-hydroxylase deficiency. Internal Medicine. 1992, 31 (6): 803-806. 10.2169/internalmedicine.31.803.
Schalkwijk J, Zweers MC, Steijlen PM: A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. The New England Journal of Medicine. 2001, 345 (16): 1167-1175. 10.1056/NEJMoa002939.
Koppens PFJ, Hoogenboezem T, Degenhart HJ: Carriership of a defective tenascin-X gene in steroid 21-hydroxylase deficiency patients: TNXB-TNXA hybrids in apparent large-scale gene conversions. Human Molecular Genetics. 2002, 11 (21): 2581-2590. 10.1093/hmg/11.21.2581.
Chen W, Kim MS, Shanbhag S: The phenotypic spectrum of contiguous deletion of CYP21A2 and tenascin XB: quadricuspid aortic valve and other midline defects. American Journal of Medical Genetics, Part A. 2009, 149 (12): 2803-2808. 10.1002/ajmg.a.33092.
Speiser PW, Serrat J, New MI, Gertner JM: Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1992, 75 (6): 1421-1424. 10.1210/jc.75.6.1421.
Sartorato P, Zulian E, Benedini S: Cardiovascular risk factors and ultrasound evaluation of intima-media thickness at common carotids, carotid bulbs, and femoral and abdominal aorta arteries in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (3): 1015-1018. 10.1210/jc.2006-1711.
Falhammar H, Filipsson H, Holmdahl G: Metabolic profile and body composition in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (1): 110-116. 10.1210/jc.2006-1350.
Dereli D, Ozgen G, Buyukkececi F, Guney E, Yilmaz C: Platelet dysfunction in lean women with polycystic ovary syndrome and association with insulin sensitivity. Journal of Clinical Endocrinology and Metabolism. 2003, 88 (5): 2263-2268. 10.1210/jc.2002-021391.
Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI: Prenatal androgenization affects gender-related behavior but not gender identity in 5–12-year-old girls with congenital adrenal hyperplasia. Archives of Sexual Behavior. 2004, 33 (2): 97-104. 10.1023/B:ASEB.0000014324.25718.51.
Berenbaum SA, Bailey JM: Effects on gender identity of prenatal androgens and genital appearance: evidence from girls with congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2003, 88 (3): 1102-1106. 10.1210/jc.2002-020782.
Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI: Sexual orientation in women with classical or non-classical congenital adrenal hyperplasia as a function of degree of prenatal androgen excess. Archives of Sexual Behavior. 2008, 37 (1): 85-99. 10.1007/s10508-007-9265-1.
Gastaud F, Bouvattier C, Duranteau L: Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (4): 1391-1396. 10.1210/jc.2006-1757.
Jääskeläinen J, Hippeläinen M, Kiekara O, Voutilainen R: Child rate, pregnancy outcome and ovarian function in females with classical 21-hydroxylase deficiency. Acta Obstetricia et Gynecologica Scandinavica. 2000, 79 (8): 687-692.
White PC: Neonatal screening for congenital adrenal hyperplasia. Nature Reviews Endocrinology. 2009, 5 (9): 490-498. 10.1038/nrendo.2009.148.
Therrell BL, Berenbaum SA, Manter-Kapanke V: Results of screening 1.9 million Texas newborns for 21-hydroxylase- deficient congenital adrenal hyperplasia. Pediatrics. 1998, 101 (4): 583-590. 10.1542/peds.101.4.583.
Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR: Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia among hyperandrogenic women: a prospective study. Fertility and Sterility. 1999, 72 (5): 915-925. 10.1016/S0015-0282(99)00383-0.
Dewailly D, Vantyghem-Haudiquet M-C, Saidsard C: Clinical and biological phenotypes in late-onset 21 hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1986, 63 (2): 418-423. 10.1210/jcem-63-2-418.
Soldin SJ, Soldin OP: Steroid hormone analysis by tandem mass spectrometry. Clinical Chemistry. 2009, 55 (6): 1061-1066. 10.1373/clinchem.2007.100008.
Janzen N, Peter M, Sander S: Newborn screening for congenital adrenal hyperplasia: additional steroid profile using liquid chromatography-tandem mass spectrometry. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (7): 2581-2589. 10.1210/jc.2006-2890.
Zeng X, Witchel SF, Dobrowolski SF, Moulder PV, Jarvik JW, Telmer CA: Detection and assignment of CYP21 mutations using peptide mass signature genotyping. Molecular Genetics and Metabolism. 2004, 82 (1): 38-47. 10.1016/j.ymgme.2004.02.006.
Fanta M, Hill M, Belácek J, Vrbíková J, Cibula D: Comparison of corticoid substitution versus combined oral contraception administration in the treatment of non-classic adrenal hyperplasia: a prospective study. Gynecological Endocrinology. 2009, 25: 398-402. 10.1080/09513590902730770.
Spritzer P, Billaud L, Thalabard J-C: Cyproterone acetate versus hydrocortisone treatment in late-onset adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 1990, 70 (3): 642-646. 10.1210/jcem-70-3-642.
Merke DP: Approach to the adult with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2008, 93 (3): 653-660. 10.1210/jc.2007-2417.
Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW: Consensus statement on 21-hydroxylase deficiency from the European society for paediatric endocrinology and the Lawson Wilkins pediatric endocrine society. Hormone Research. 2002, 58: 188-195. 10.1159/000065490.
Azziz R, Koulianos G: Adrenal androgens and reproductive aging in females. Seminars in Reproductive Endocrinology. 1991, 9 (3): 249-260. 10.1055/s-2007-1019416.
David M, Forest MG: Prenatal treatment of congenital adrenal hyperplasia resulting from 21-hydroxylase deficiency. Journal of Pediatrics. 1984, 105 (5): 799-803. 10.1016/S0022-3476(84)80310-8.
New MI, Carlson A, Obeid J: Prenatal diagnosis for congenital adrenal hyperplasia in 532 pregnancies. Journal of Clinical Endocrinology and Metabolism. 2001, 86 (12): 5651-5657. 10.1210/jc.86.12.5651.
Pang S, Clark AT, Freeman LC: Maternal side effects of prenatal dexamethasone therapy for fetal congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 1992, 75 (1): 249-253. 10.1210/jc.75.1.249.
Miller WL: Dexamethasone treatment of congenital adrenal hyperplasia in utero: an experimental therapy of unproven safety. Journal of Urology. 1999, 162 (2): 537-540. 10.1016/S0022-5347(05)68624-7.
Speiser PW: Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine clinical practice guideline. Journal of Clinical Endocrinology and Metabolism. In press
 -hydroxysteroid dehydrogenase deficiency. Journal of Clinical Endocrinology and Metabolism. 2002, 87 (6): 2611-2622. 10.1210/jc.87.6.2611.
-hydroxysteroid dehydrogenase deficiency. Journal of Clinical Endocrinology and Metabolism. 2002, 87 (6): 2611-2622. 10.1210/jc.87.6.2611.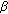 - hydroxylase deficiency. Human Molecular Genetics. 1997, 6 (11): 1829-1834. 10.1093/hmg/6.11.1829.
- hydroxylase deficiency. Human Molecular Genetics. 1997, 6 (11): 1829-1834. 10.1093/hmg/6.11.1829.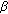 -Hydroxysteroid dehydrogenase deficiency in hyperandrogenism. American Journal of Obstetrics and Gynecology. 1993, 168 (3): 889-895.
-Hydroxysteroid dehydrogenase deficiency in hyperandrogenism. American Journal of Obstetrics and Gynecology. 1993, 168 (3): 889-895.Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Witchel, S.F., Azziz, R. Nonclassic Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol 2010, 625105 (2010). https://doi.org/10.1155/2010/625105
Received:
Accepted:
Published:
DOI: https://doi.org/10.1155/2010/625105