- Review Article
- Open access
- Published:
Mineralocorticoid Deficiency and Treatment in Congenital Adrenal Hyperplasia
International Journal of Pediatric Endocrinology volume 2010, Article number: 656925 (2010)
Abstract
Approximately 75%–80% of patients with Congenital Adrenal Hyperplasia (CAH) fail to synthesize sufficient mineralocorticoids to maintain salt and water balance. In most instances genotype can predict mineralocorticoid deficiency in CAH. Early recognition and replacement with 9 -fludrocortisone and salt supplements will prevent development of potentially lethal salt losing crises. In infancy a relative state of aldosterone resistance exists and replacement dose of 9
-fludrocortisone and salt supplements will prevent development of potentially lethal salt losing crises. In infancy a relative state of aldosterone resistance exists and replacement dose of 9 -fludrocortisone based on body surface area is higher during infancy compared to childhood and adults. Salt supplementation is generally not required after weaning is started. Regular monitoring of blood pressure and measurements of plasma electrolytes and renin are required to prevent complications of under or over dosage.
-fludrocortisone based on body surface area is higher during infancy compared to childhood and adults. Salt supplementation is generally not required after weaning is started. Regular monitoring of blood pressure and measurements of plasma electrolytes and renin are required to prevent complications of under or over dosage.
1. Introduction
Congenital adrenal hyperplasia (CAH) is a disorder of steroidogenesis in which cortisol metabolism primarily is impaired. Mineralocorticoid deficiency in CAH is a feature of 21-Hydroxylase (21-OHD), 3 -Hydroxysteroid dehydrogenase (3
-Hydroxysteroid dehydrogenase (3 -HSD) deficiencies and lipoid adrenal hyperplasia. Deficiency of 21-OHD accounts for 90% of cases of CAH [1] while deficiency of 3
-HSD) deficiencies and lipoid adrenal hyperplasia. Deficiency of 21-OHD accounts for 90% of cases of CAH [1] while deficiency of 3 -HSD, and lipoid adrenal hyperplasia are rare causes of CAH. Approximately 75% of cases of 21-OHD [1] and 60%–70% of cases of 3
-HSD, and lipoid adrenal hyperplasia are rare causes of CAH. Approximately 75% of cases of 21-OHD [1] and 60%–70% of cases of 3 -HSD deficiency [2] cannot synthesise sufficient aldosterone and are classed as "salt wasters."
-HSD deficiency [2] cannot synthesise sufficient aldosterone and are classed as "salt wasters."
2. Genotype-Phenotype Correlation of Aldosterone Deficiency
Several studies have demonstrated strong genetic association between the degree of mineralocorticoid and glucocorticoid deficiency in 21-OHD CAH and abnormalities in the CYP 21 gene [3–6]. Large gene deletions and point mutations which are expected to produce no measurable enzyme activity are associated with aldosterone deficiency and salt-wasting 21-OHD [7]. Exceptions however exist and a similar mutation can be associated with more than one phenotype giving rise to genotype-phenotype discordance [5]. The I172N CYP21 mutation is mainly associated with simple virilizing CAH but can also cause salt wasting CAH [8]. A significant difference exists between the degree of genotype-phenotype correlation demonstrated in relation to the concentration of aldosterone and that of genital phenotype [9]. The relationship with degree of virilization with genotype and aldosterone concentration is less predictable as various genetic factors regulate androgen biosynthesis and sensitivity or the ability to retain sodium through extra-adrenal mechanisms [9, 10]. A degree and spectrum of aldosterone deficiency and salt loss exists in all forms of CAH including those with simple virilising and nonclassical forms of CAH [2, 11, 12]. In clinical practice this is only relevant in very hot countries where extreme salt deprivation can manifest.
Glucocorticoids are essential for development and function of adrenal medulla and adrenomedullary function is compromised in CAH [13]. Plasma metanephrine concentration is significantly low in severe salt wasting CAH compared to simple virilizing form of CAH and is comparable to genotype in predicting aldosterone deficiency [14]. Combination of plasma metanephrine and genotype can increase the predictability of phenotype in CAH.
Genotype phenotype association is much stronger in patients with 3 -HSD deficiency with most studies demonstrating more precise relationship with genotype and activity of aldosterone [15].
-HSD deficiency with most studies demonstrating more precise relationship with genotype and activity of aldosterone [15].
3. Aldosterone Biosynthesis and Physiological Role in Salt and Water Balance (Figure 1)
Aldosterone is synthesized in the zona glomerulosa of the adrenal cortex from corticosterone by aldosterone synthetase (CYP11B2). The rate of aldosterone synthesis is 100–1000 fold less than cortisol. While cortisol circulates in the serum in nanomolar concentration, the concentration of aldosterone is 1000-fold less. Aldosterone secretion is mainly regulated by serum potassium and angiotensin II, with ACTH having only a short term effect [15].
Renin is secreted by the juxtaglomerular apparatus in response to decreased intravascular volume (Figure 1). Renin cleaves angiotensinogen into angiotensin I. Angiotensin-converting enzyme in the lungs cleaves angiotensin I to angiotensin II, a potent vasopressor agent. Angiotensin II acts through the angiotensin II receptor to hydrolyze phosphatidylinositol bisphosphate to inositol triphosphate and diacylglycerol. These raise intracellular calcium concentrations and activate protein kinase C and calmodulin-activated (CaM) kinases. Increased concentration of extracellular potassium depolarizes the cell membrane and increases calcium influx through voltage-gated calcium channels which similarly activate protein kinase C and calmodulin-activated (CaM) kinases. Phosphorylation of transcriptional regulatory factors by CaM kinases increases transcription of the aldosterone synthase (CYP11B2) gene required for aldosterone synthesis.

Aldosterone biosynthesis through renin-angiotensin system.
4. Ontogeny of Aldosterone Secretion
The renin-angiotensin-aldosterone system (RAAS) is known to be active by around 16 weeks of gestational age [16]. In the neonatal period a relative state of aldosterone resistance exists. In extreme prematurity aldosterone concentration can be as high as 12–29,000 pmol/L [17]. In term neonates the average concentration of aldosterone is 22,000 pmol/L [18] (upper limit of normal range in adult is 970 pmol/L [17]). During the first year of age, there is a steep, progressive decline in aldosterone to prepubertal concentrations [19]. The physiological basis for the relative aldosterone resistance in the neonatal period is unclear. Renal tubular immaturity leading to sodium loss could trigger the activation of RAAS alternatively impaired ability of renal tubules to respond to aldosterone action may constitute a protective mechanism against the high plasma concentration of aldosterone [18].
It is not uncommon in CAH to encounter a normal or even higher concentration of aldosterone in neonates (compared to childhood range) due to physiological resistance and possible assay interference from circulating steroids. The clue to diagnosis in these situations is the rising plasma potassium concentration which precedes the onset of hyponatremia around day 7–10 of postnatal life. These changes are associated with an increase in plasma renin activity and if left unchecked the development of a salt-losing crisis. In a recent study in healthy term neonates Martinerie et al. documented a poor correlation of serum aldosterone with serum potassium while urinary aldosterone appeared to perform better. These data if replicated suggest that spot urinary measurement of aldosterone/creatinine concentration along with plasma and urine electrolytes could be a more sensitive and better index for evaluation of mineralocorticoid effector mechanisms [18].
5. Management of Salt Loss in CAH
Most of the patients with aldosterone deficiency present in early infancy with salt loss which can be fatal if not identified and treated early. Salt loss crises classically manifest at the end of the first week of life. In girls the ambiguity of the genitalia alerts the clinician to a diagnosis of CAH and associated aldosterone deficiency. Male infants present with increasing virilization and increased pigmentation of scrotum but this can be difficult to identify in early infancy which raises the chance that the actual initial presentation will be with a salt-losing crisis. Not all infants present with a salt-losing crisis and failure to thrive is another form of presentation.
Aldosterone deficiency results in excessive sodium and insufficient potassium excretion in the urine which results in hyperkalemia, hyponatremia, hypovolemia, and hyperreninemia. Cortisol deficiency in these patients contributes to hypoglycemia, poor cardiac function, poor vascular response to catecholamines, a decreased glomerular filtration rate, and increased secretion of antidiuretic hormone [20]. Cortisol and aldosterone deficiency together cause hyponatremic dehydration and shock. Shock is further exacerbated by the deficiency of catecholamines which often accompanies salt-losing CAH [13].
The acute circulatory collapse needs intravenous resuscitation with normal saline, glucose, and intravenous hydrocortisone. In females with ambiguous genitalia and males with a family history of CAH the acute salt-losing state can be avoided by daily monitoring of the serum electrolyte concentrations in the neonatal period. The first change is an increase in the serum potassium concentration above 5.5 mmol/L and this may be associated with a serum sodium concentration in the lower quartile of the normal range or actual hyponatremia. At this point blood should be drawn for the measurement of aldosterone and plasma renin activity.
Treatment will probably be already ongoing with oral hydrocortisone (10–12 mg/m2/day,  3–4.5 mg/day) and 9
3–4.5 mg/day) and 9 -fludrocortisone (150
-fludrocortisone (150  g/m2/day,
g/m2/day,  50
50  g/day) with sodium supplements of up to 10–12 mmol/Kg/day (Sodium 1 mmol
g/day) with sodium supplements of up to 10–12 mmol/Kg/day (Sodium 1 mmol  1 mEq) should be introduced. If a salt-losing crisis does not manifest in the 7–10 days of life, ongoing monitoring with twice weekly electrolytes and weekly measures of weight should be undertaken out to about 6–8 weeks of life.
1 mEq) should be introduced. If a salt-losing crisis does not manifest in the 7–10 days of life, ongoing monitoring with twice weekly electrolytes and weekly measures of weight should be undertaken out to about 6–8 weeks of life.
Salt supplementation is required during infancy as the daily intake of milk (breast or milk formula) provides only 2 mmol/kg/day of sodium which is insufficient to replace the sodium deficit that arises in CAH. Unless there is adequate replacement of sodium simply increasing the dose of 9 -fludrocortisone will not normalize the serum sodium. Some clinicians use higher doses of 9
-fludrocortisone will not normalize the serum sodium. Some clinicians use higher doses of 9 -fludrocortisone up to 350
-fludrocortisone up to 350  g/day [21] but the authors find that in the majority of cases a 9
g/day [21] but the authors find that in the majority of cases a 9 -fludrocortisone dose of 150
-fludrocortisone dose of 150  g/m2/day (
g/m2/day ( 50
50  g/day) with salt supplements of 5–8 mmol/kg/day is sufficient. In the event of persistent hyponatremia dose of 9
g/day) with salt supplements of 5–8 mmol/kg/day is sufficient. In the event of persistent hyponatremia dose of 9 -fludrocortisone is increased only after 10–12 mmol/kg/day of sodium supplements have failed to normalize the serum sodium. Care needs to be taken with high doses of 9
-fludrocortisone is increased only after 10–12 mmol/kg/day of sodium supplements have failed to normalize the serum sodium. Care needs to be taken with high doses of 9 -fludrocortisone as the medication has dexamethasone like glucocorticoid potency and any dose of 9α-fludrocortisone needs to be factored into the calculation of the total glucocorticoid daily dose. Once the serum sodium normalizes monitoring of serum electrolytes, blood pressure is required to titrate the dose of sodium and 9α-fludrocortisone supplements. Once weaning is fully established, by around 6–8 months of age, salt supplements are generally not required.
-fludrocortisone as the medication has dexamethasone like glucocorticoid potency and any dose of 9α-fludrocortisone needs to be factored into the calculation of the total glucocorticoid daily dose. Once the serum sodium normalizes monitoring of serum electrolytes, blood pressure is required to titrate the dose of sodium and 9α-fludrocortisone supplements. Once weaning is fully established, by around 6–8 months of age, salt supplements are generally not required.
The dose of 9α-fludrocortisone during the first and second years of life in CAH is higher than that at other stages of life. At the point of the salt-losing crisis the requirement for a higher dose of 9α-fludrocortisone probably results from the anti-aldosterone nature of the precursors accumulated in 21-hydroxylase deficiency [22]. As the child grows, a maximum dose of 100  g/day is usually sufficient and in adolescent and adults 50–100
g/day is usually sufficient and in adolescent and adults 50–100  g/day is the usual requirement [23].
g/day is the usual requirement [23].
The half life of aldosterone is relatively short; however 9 -fludrocortisone has a longer duration of action and can usually be administered once a day although twice daily therapy with two equally divided doses is practiced [21]. This can be especially valuable approach in situations of increased physiological loss of fluid and electrolytes, for example, heat, when patients may require higher and more frequent doses of 9
-fludrocortisone has a longer duration of action and can usually be administered once a day although twice daily therapy with two equally divided doses is practiced [21]. This can be especially valuable approach in situations of increased physiological loss of fluid and electrolytes, for example, heat, when patients may require higher and more frequent doses of 9 -fludrocortisone [24].
-fludrocortisone [24].
CAH and its management is complicated by obesity, insulin resistance [25–27], and elevated 24-hour ambulatory arterial blood pressure measurements with absence of the nocturnal dip in blood pressure [28]. These complications place patients at significant risk for developing metabolic syndrome-related atherosclerotic cardiovascular disease in adult life [28]. For future cardiovascular health it is of paramount importance to keep the serum electrolytes and blood pressure within the normal range with the right balance of sodium supplements and 9 -fludrocortisone replacement. Overtreatment can lead to hypertension which can go unnoticed as children largely remain asymptomatic. With dexamethasone-like glucocorticoid potency of 9
-fludrocortisone replacement. Overtreatment can lead to hypertension which can go unnoticed as children largely remain asymptomatic. With dexamethasone-like glucocorticoid potency of 9 -fludrocortisone, overtreatment can also impact on growth and body composition and impart insulin resistance. During the first year of life treatment with 9
-fludrocortisone, overtreatment can also impact on growth and body composition and impart insulin resistance. During the first year of life treatment with 9 -fludrocortisone is monitored by measurement of serum electrolytes along with blood pressure every 2-3 months. During the second year of life monitoring of treatment can be performed every 6 months followed by yearly in childhood and adolescence.
-fludrocortisone is monitored by measurement of serum electrolytes along with blood pressure every 2-3 months. During the second year of life monitoring of treatment can be performed every 6 months followed by yearly in childhood and adolescence.
For the dose at which 9 -fludrocortisone is administered, drug interactions are minimal. However, caution must be taken when it is concomitantly used with antihypertensives especially those which act through RAAS pathway. 9α-fludrocortisone can potentiate the hypokalemic effects of sympathomimetics, Beta2 agonist, and theophylline especially when used with IV hydrocortisone in patients with acute severe asthma.
-fludrocortisone is administered, drug interactions are minimal. However, caution must be taken when it is concomitantly used with antihypertensives especially those which act through RAAS pathway. 9α-fludrocortisone can potentiate the hypokalemic effects of sympathomimetics, Beta2 agonist, and theophylline especially when used with IV hydrocortisone in patients with acute severe asthma.
The need for extra mineralocorticoid replacement during stress and elective surgery is unclear. Where oral intake is possible 9 -fludrocortisone should be administered. If oral intake is not possible, the doses of intravenous hydrocortisone recommended for stressful conditions [23] often circumvent the need for 9
-fludrocortisone should be administered. If oral intake is not possible, the doses of intravenous hydrocortisone recommended for stressful conditions [23] often circumvent the need for 9 -fludrocortisone as the mineralocorticoid potency of 20 mg hydrocortisone is equivalent to 50
-fludrocortisone as the mineralocorticoid potency of 20 mg hydrocortisone is equivalent to 50  g of 9
g of 9 -fludrocortisone. Regular monitoring of serum and urine electrolytes should be undertaken with adequate intravenous substitution of sodium. Oral 9α-fludrocortisone should be commenced as soon as oral intake is feasible and gastrointestinal absorption is deemed possible.
-fludrocortisone. Regular monitoring of serum and urine electrolytes should be undertaken with adequate intravenous substitution of sodium. Oral 9α-fludrocortisone should be commenced as soon as oral intake is feasible and gastrointestinal absorption is deemed possible.
6. Summary: Practice Points
-
(1)
Approximately 75%–80% of patients with CAH fail to synthesize sufficient aldosterone to maintain salt balance.
-
(2)
Early recognition and replacement with 9
 -fludrocortisone and salt supplements will prevent development of potentially lethal salt losing crises.
-fludrocortisone and salt supplements will prevent development of potentially lethal salt losing crises. -
(3)
Salt supplementation is generally not required after weaning is started.
-
(4)
Dose of aldosterone is traditionally calculated based on body surface area and is higher during infancy compared to childhood and adults.
-
(5)
Regular monitoring of blood pressure and measurement of plasma electrolytes and renin are required to prevent complications of under or over dosage.
 -fludrocortisone and salt supplements will prevent development of potentially lethal salt losing crises.
-fludrocortisone and salt supplements will prevent development of potentially lethal salt losing crises.References
White PC, Speiser PW: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrinology Reviews. 2000, 21 (3): 245-291. 10.1210/er.21.3.245.
Rhéaume E, Simard J, Morel Y: Congenital adrenal hyperplasia due to point mutations in the type II 3
 -hydroxysteroid dehydrogenase gene. Nature Genetics. 1992, 1 (4): 239-245. 10.1038/ng0792-239.
-hydroxysteroid dehydrogenase gene. Nature Genetics. 1992, 1 (4): 239-245. 10.1038/ng0792-239.Jääskeläinen J, Levo A, Voutilainen R, Partanen J: Population-wide evaluation of disease manifestation in relation to molecular genotype in steroid 21-hydroxylase (CYP21) deficiency: good correlation in a well defined population. Journal of Clinical Endocrinology & Metabolism. 1997, 82 (10): 3293-3297. 10.1210/jc.82.10.3293.
Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP: Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. Journal of Clinical Endocrinology & Metabolism. 2000, 85 (3): 1059-1065. 10.1210/jc.85.3.1059.
Speiser PW, Dupont J, Zhu D: Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Investigation. 1992, 90 (2): 584-595. 10.1172/JCI115897.
Wedell A, Thilen A, Ritzen EM, Stengler B, Luthman H: Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. Journal of Clinical Endocrinology & Metabolism. 1994, 78 (5): 1145-1152. 10.1210/jc.78.5.1145.
White PC, New MI, Dupont B: HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proceedings of the National Academy of Sciences of the United States of America. 1984, 81 (23): 7505-7509. 10.1073/pnas.81.23.7505.
Wilson RC, Mercado AB, Cheng KC, New MI: Steroid 21-hydroxylase deficiency: genotype may not predict phenotype. Journal of Clinical Endocrinology & Metabolism. 1995, 80 (8): 2322-2329. 10.1210/jc.80.8.2322.
Riepe FG, Sippell WG: Recent advances in diagnosis, treatment, and outcome of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Reviews in Endocrine and Metabolic Disorders. 2007, 8 (4): 349-363. 10.1007/s11154-007-9053-1.
Huynh T, McGown I, Cowley D: The clinical and biochemical spectrum of congenital adrenal hyperplasia secondary to 21-hydroxylase deficiency. The Clinical Biochemistry Reviews. 2009, 30 (2): 75-86.
Grant DB, Dillon MJ, Atherden SM, Levinsky RJ: Congenital adrenal hyperplasia: renin and steroid values during treatment. European Journal of Pediatrics. 1977, 126 (1-2): 89-96. 10.1007/BF00443127.
Nimkarn S, Lin-Su K, Berglind N, Wilson RC, New MI: Aldosterone-to-renin ratio as a marker for disease severity in 21-hydroxylase deficiency congenital adrenal hyperplasia. Journal of Clinical Endocrinology & Metabolism. 2007, 92 (1): 137-142. 10.1210/jc.2006-0964.
Merke DP, Chrousos GP, Eisenhofer G: Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. The New England Journal of Medicine. 2000, 343 (19): 1362-1368. 10.1056/NEJM200011093431903.
Charmandari E, Eisenhofer G, Mehlinger SL: Adrenomedullary function may predict phenotype and genotype in classic 21-hydroxylase deficiency. Journal of Clinical Endocrinology & Metabolism. 2002, 87 (7): 3031-3037. 10.1210/jc.87.7.3031.
Simard J, Ricketts M-L, Gingras S, Soucy P, Feltus FA, Melner MH: Molecular biology of the 3
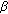 -hydroxysteroid dehydrogenase/
-hydroxysteroid dehydrogenase/ 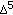 -
-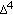 isomerase gene family. Endocrine Reviews. 2005, 26 (4): 525-582. 10.1210/er.2002-0050.
isomerase gene family. Endocrine Reviews. 2005, 26 (4): 525-582. 10.1210/er.2002-0050.Partsch C-J, Sippell WG, Mackenzie IZ, Aynsley-Green A: The steroid hormonal milieu of the undisturbed human fetus and mother at 16–20 weeks gestation. Journal of Clinical Endocrinology & Metabolism. 1991, 73 (5): 969-974. 10.1210/jcem-73-5-969.
Bourchier D: Plasma aldosterone levels in the 1st week of life in infants of less than 30 weeks gestation. European Journal of Pediatrics. 2005, 164 (3): 141-145. 10.1007/s00431-004-1572-0.
Martinerie L, Pussard E, Foix-L'Hélias L: Physiological partial aldosterone resistance in human newborns. Pediatric Research. 2009, 66 (3): 323-328. 10.1203/PDR.0b013e3181b1bbec.
Sippell WG, Doerr HG, Bidlingmaier F, Knorr D: Plasma levels of aldosterone, corticosterone, 11-deoxycorticosterone, progesterone, 17-hydroxyprogesterone, cortisol, and cortisone during infancy and childhood. Pediatric Research. 1980, 14 (1): 39-46.
Lamberts SWJ, Bruining HA, de Jong FH: Corticosteroid therapy in severe illness. The New England Journal of Medicine. 1997, 337 (18): 1285-1292. 10.1056/NEJM199710303371807.
Riepe FG, Krone N, Viemann M, Partsch C-J, Sippell WG: Management of congenital adrenal hyperplasia: results of the ESPE questionnaire. Hormone Research. 2002, 58 (4): 196-205. 10.1159/000065492.
Oelkers WKH: Effects of estrogens and progestogens on the renin-aldosterone system and blood pressure. Steroids. 1996, 61 (4): 166-171. 10.1016/0039-128X(96)00007-4.
Hindmarsh PC: Management of the child with congenital adrenal hyperplasia. Best Practice & Research Clinical Endocrinology & Metabolism. 2009, 23 (2): 193-208. 10.1016/j.beem.2008.10.010.
Hahner S, Allolio B: Therapeutic management of adrenal insufficiency. Best Practice & Research Clinical Endocrinology & Metabolism. 2009, 23 (2): 167-179. 10.1016/j.beem.2008.09.009.
Charmandari E, Weise M, Bornstein SR: Children with classic congenital adrenal hyperplasia have elevated serum leptin concentrations and insulin resistance: potential clinical implications. Journal of Clinical Endocrinology & Metabolism. 2002, 87 (5): 2114-2120. 10.1210/jc.87.5.2114.
Völkl TMK, Simm D, Beier C, Dörr HG: Obesity among children and adolescents with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics. 2006, 117 (1): e98-e105. 10.1542/peds.2005-1005.
Williams RM, Deeb A, Ong KK: Insulin sensitivity and body composition in children with classical and nonclassical congenital adrenal hyperplasia. Clinical Endocrinology. 2010, 72 (2): 155-160. 10.1111/j.1365-2265.2009.03587.x.
Roche EF, Charmandari E, Dattani MT, Hindmarsh PC: Blood pressure in children and adolescents with congenital adrenal hyperplasia (21-hydroxylase deficiency): a preliminary report. Clinical Endocrinology. 2003, 58 (5): 589-596. 10.1046/j.1365-2265.2003.01757.x.
 -hydroxysteroid dehydrogenase gene. Nature Genetics. 1992, 1 (4): 239-245. 10.1038/ng0792-239.
-hydroxysteroid dehydrogenase gene. Nature Genetics. 1992, 1 (4): 239-245. 10.1038/ng0792-239.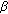 -hydroxysteroid dehydrogenase/
-hydroxysteroid dehydrogenase/ 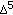 -
-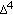 isomerase gene family. Endocrine Reviews. 2005, 26 (4): 525-582. 10.1210/er.2002-0050.
isomerase gene family. Endocrine Reviews. 2005, 26 (4): 525-582. 10.1210/er.2002-0050.Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Padidela, R., Hindmarsh, P.C. Mineralocorticoid Deficiency and Treatment in Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol 2010, 656925 (2010). https://doi.org/10.1155/2010/656925
Received:
Accepted:
Published:
DOI: https://doi.org/10.1155/2010/656925