- Review Article
- Open access
- Published:
Management of the Adult with Congenital Adrenal Hyperplasia
International Journal of Pediatric Endocrinology volume 2010, Article number: 614107 (2010)
Abstract
Congenital adrenal hyperplasia (CAH), most commonly due to 21-hydroxylase deficiency (21OHD), has been studied by pediatric endocrinologists for decades. Advances in the care of these patients have enabled many of these children to reach adulthood. In contrast to the course and management of the disease in childhood, little is known about CAH in adults. In many patients, the proclivity to salt-wasting crises decreases. Linear growth ceases, and reproductive function becomes an issue. Most importantly, management must minimize the potential for long-term consequences of conventional therapies. Here we review the existing literature regarding comorbidities of adults with 21OHD, goals of treatment, and approaches to therapy, with an emphasis on need for improved management strategies.
1. Introduction
As discussed elsewhere, the congenital adrenal hyperplasias (CAHs) are genetic defects in cortisol biosynthesis. Low cortisol removes feedback inhibition of adrenocorticotropin (ACTH) secretion, which causes adrenal hyperplasia. The clinical consequences of CAH derive from both the shunting of cortisol precursors along other pathways and the biological activities of these precursors and their unusual metabolites, which accumulate above the block. Treatments will ideally replace the glucocorticoid deficiency and normalize both mineralocorticoid and androgen biosynthesis without inducing untoward effects from the drugs themselves. The most common cause of CAH is 21-hydroxylase deficiency (21-OHD) [1], which afflicts about 1 : 15,000 live births [2]. Since the introduction of cortisone therapy by Wilkins et al. in the early 1950s [3], these children have been able to survive into adulthood. Now that over a half century has passed, one would think that abundant information would have accumulated on the care of adults with CAH, as is now the case for children [4]. Regrettably, very little is known about the physiology and management of adults with CAH, and what is known is essentially limited to 21-OHD.
1.1. Why Is So Little Known?
Genetic disorders, which manifest with congenital disease, are largely the providence of pediatrics. With the completion of the Human Genome Project, internists have become more aware of genetic disorders, but largely the focus has been on susceptibility genes for cancer, diabetes, and cardiovascular diseases. Training in the care of patients with congenital biosynthetic defects, such as glycogen storage diseases and CAH, is generally not considered an important component of internal medicine residencies and endocrinology fellowships.
Consequently, few doctors who care for adults, even those at academic medical centers, are adequately trained or interested in rare genetic diseases. This scenario is evidently the case for CAH. Most internal medicine endocrinology trainees will see only a few patients with CAH, mainly if they rotate in the pediatric endocrinology clinic, and many will never see a single adult with CAH during their training. A search of the NIH CRISP database revealed only a few grants awarded to the study of CAH in the last 5 years, none of which were awarded to investigators in departments of internal medicine. Without interest and research in academic centers, there is little chance that internal medicine endocrinology fellows will receive adequate training in CAH.
1.2. Children Are Not Little Adults
The endocrine physiology of childhood is dominated by growth and pubertal development. Adults do neither, but they do age, and many have children or at least wish to become parents. With age, they are prone to all the maladies of adult life, including heart disease, osteoporosis, and cancer. Consequently, the focus and goals of treatment are quite different in adults and children. Treatment of CAH in infancy and early childhood strives first to prevent salt-wasting and hypotensive crises due to adrenal insufficiency. Treatment of adults with CAH should be tailored to meet the needs of the patient at the present time, but with a long-term view of mitigating consequences of therapy. As a rule, the medications used and intensity of monitoring will vary as the objectives change with time [5].
1.3. Young Adults with Chronic Diseases Are Weary of Seeing Doctors
This feeling is particularly true if the doctor knows very little about their condition and shows little interest or concern for their specific needs. Many patients with CAH have stopped seeing physicians altogether and have discontinued corticosteroid replacement for long periods of time [6]. Women may become comfortable living in a state of androgen excess and may even experience fatigue from testosterone withdrawal if therapy is resumed. Men with CAH of experience few symptoms from reducing or stopping therapy, until they become seriously ill or their testicular rests become uncomfortably large. These considerations are important in understanding the approach to the adult with CAH, both medically and psychologically.
2. Physiology of CAH in Adults
Many of the general principles are the same as for children with CAH, but the importance of the various factors is considerably different. All subsequent discussion will be limited to 21-OHD.
2.1. Basic Adrenal Physiology
Cytochrome P450c21 (CYP21A2) deficiency precludes aldosterone and cortisol synthesis, limiting steroidogenesis to the reactions catalyzed by 3β-hydroxysteroid dehydrogenase type 2 (3β-HSD2), cytochrome P450c17 (CYP17A1), and a bit to cytochrome P450c11β (CYP11B1) (Figure 1(a)). Low cortisol increases ACTH production, flooding the adrenal steroidogenic machinery with upstream precursors (Figure 1(b)). Over time, the adrenals enlarge due to the chronic trophic stimulus of ACTH. Consequently, the adult adrenal of a patient with 21-OHD makes large amounts of a few steroids, mainly progesterone (P4), 17-hydroxyprogesterone (17-OHP), dehydroepiandrosterone and its sulfate (DHEA[S]), plus lesser amounts of androstenedione (AD), testosterone (T) [7], and 21-deoxycortisol—which has little glucocorticoid or mineralocorticoid activity.

Steroid biosynthesis in 21-OHD. (a): Normal steroid biosynthetic pathways within each zone of the human adrenal gland. The dashed arrows indicate minor reactions. (b): Altered steroid biosynthesis in 21-OHD. The block (heavy vertical line) shunts precursors to 19-carbon steroids (heavy curved black arrow at left).
Due to zonation, two critical enzyme activities required for T synthesis are physically separated in the adrenal gland. The zona fasciculata cells, which contain 3β-HSD2 activity, also contain CYP17A1; however, these cells have low 17,20-lyase activity because expression of the important cofactor protein cytochrome  (b5) is low [8, 9]. In contrast, the zona reticularis—in which ACTH also stimulates steroid production—has abundant CYP17A1, b5, and thus high 17,20-lyase activity, but these cells are deficient in 3β-HSD2 and have robust DHEA-sulfotransferase activity (SULT2A1). Furthermore, the adrenal gland lacks 17β-HSD3, the enzyme responsible for reduction of AD to T in the testis, but contains AKR1C3 (17β-HSD5) [7], which has weak activity in catalyzing this reaction [10]. Consequently, the adrenal gland of a patient with 21-OHD has high capacity to synthesize DHEAS, but little of this 19-carbon precursor escapes the adrenal as T.
(b5) is low [8, 9]. In contrast, the zona reticularis—in which ACTH also stimulates steroid production—has abundant CYP17A1, b5, and thus high 17,20-lyase activity, but these cells are deficient in 3β-HSD2 and have robust DHEA-sulfotransferase activity (SULT2A1). Furthermore, the adrenal gland lacks 17β-HSD3, the enzyme responsible for reduction of AD to T in the testis, but contains AKR1C3 (17β-HSD5) [7], which has weak activity in catalyzing this reaction [10]. Consequently, the adrenal gland of a patient with 21-OHD has high capacity to synthesize DHEAS, but little of this 19-carbon precursor escapes the adrenal as T.
Alternative routes to 5α-reduced, 19-carbon steroids have been described in other species [11, 12]. These routes involve the 5α-reduction of 21-carbon pregnanes P4 and 17-OHP, followed by 3α-reduction, to yield 5α-pregnan-3α-ol-20-one (allopregnanolone) and its 17α-hydroxy derivative, 5α-pregnane-3α, 17α-diol-20-one (Pdiol). Pdiol is then cleaved to androsterone by the 17,20-lyase activity of CYP17A1. Androsterone reduction by 17β-HSD enzymes yields 5α-androstane-3α, 17β-diol (Adiol), which is metabolized by oxidative 3α-HSD enzymes such as 17β-HSD6 (RODH) [13] to DHT (Figure 2(b)). Via this pathway, DHT is formed without the intermediacy of T or AD [14]. Considerable genetic and biochemical evidence suggests that this route contributes to DHT formation and genital virilization in female fetuses with cytochrome P450-oxidoreductase deficiency, presumably due to the accumulation of 17-OHP [15–18]. In 21-OHD, intra-adrenal 17-OHP accumulates as well; consequently, this pathway is likely to be important in the fetus with 21-OHD [19]. In adults with 21-OHD, however, the pathway may not be very active, due to lower expression of 5α-reductase activity in the adult adrenal.

Alternate and peripheral androgen synthesis pathways. (a): the alternate or "backdoor" pathway to DHT, with intra- and extra-adrenal compartments demarcated. (b): simplified diagram of peripheral androgen and estrogen metabolism.
2.2. Extra-Adrenal Steroid Metabolism
Based on the above discussion, the adrenal glands of adults with 21-OHD produce little T and huge amounts of other steroids, yet none are active hormones except for P4. Nevertheless, without proper treatment, AD, T, and DHT concentrations are high in women and men with 21-OHD, and a high proportion of T in men with 21-OHD derives from the adrenal. This T is produced by peripheral metabolism of the adrenal secretions, primarily in the liver but also in skin and even target tissues. The peripheral metabolism of steroids is more difficult to define than the intra-adrenal steroidogenic pathways, due to redundancy of enzymes, competing routes of metabolism, and variability amongst individuals. Nevertheless, steroid sulfatase (STS) is present in liver, skin, and many other tissues [20], and these tissues also contain 3β-HSD type 1 [21]. Conversion of AD to T is efficiently catalyzed by 17β-HSD3, but its cognate gene is only expressed in the Leydig cell of the testis. As mentioned above, 17β-HSD5 also has weak activity for conversion of AD to T [10], but other routes from DHEA to T also exist. DHEA can be reduced by 17β-HSD1, which is an efficient enzyme [22], to androst-5-ene-3β, 17β-diol, a substrate for 3β-HSDs, yielding T (Figure 2(a)). Even 17β-HSD2, which is thought of as an "inactivating'' enzyme by efficiently converting T to AD [23], achieves a pseudo-equilibrium in intact cells with 1-2% T generated from AD and increases the apparent potency of AD as an androgen in model systems [24]. In liver and skin, 5α-reductase type 1 [25] further activates T to DHT, but T and DHT are also substrates for UDP-glucuronyl transferases (UGTs), which conjugate active androgens and their metabolites for excretion in the urine [26]. Consequently, the route(s) from DHEA to T even in normal women are not obvious, and the biochemistry is even more complex in 21-OHD, as androgen precursor production is very high.
In addition to peripheral conversion of DHEA[S] to active androgens, P4, which accumulates 2 steps prior to the block, can be 21-hydroxylated by hepatic cytochromes P450 [27, 28] to yield 11-deoxycorticosterone (DOC), a potent mineralocorticoid. The extent and routes of peripheral metabolism vary considerably amongst individuals and might explain to some degree the differences in disease control for adults with 21-OHD. Conversely, greater accumulation of P4 can contribute to anovulation and infertility in women with 21-OHD. For these reasons, not only the control of steroids exiting the adrenal, but also the extra-adrenal metabolism of these steroids is an important consideration in understanding the physiology of CAH in the adult.
3. Management of CAH in Adults
As with physiology, the principles governing the treatment of adults with CAH share many similarities with those for children with CAH; however, the differences relate to growth, pubertal development, and fertility. In addition, the care of adults is often more individualized based on patient choices than in children.
3.1. General Approach to the Patient
As previously mentioned, the treatment goals of patients with 21-OHD evolve with the transition to adulthood. Salt-wasting crises occur much less frequently, typically only during a severe concurrent illness. During childhood, tight control prevents premature somatic growth and development of secondary sexual characteristics, but both concerns are no longer germane in adulthood. The three major concerns for the adult with 21-OHD are: (1) the prevention of adrenal and gonadal hyperplasia and neoplasia; (2) the prevention of long-term consequences of adrenal replacement therapies, and (3) the restoration of fertility in those who desire to have children. Consequently, the intensity and complexity of treatment regimen is individualized and changed over time to meet the needs of the patient and to optimize the risk/benefit ratio.
Mineralocorticoid replacement with 9α-fludrocortisone acetate, 0.05–0.2 mg/d, is usually continued, with the goal of a suppressed or low-normal plasma renin activity, which blunts the hypovolemic drive to ACTH production. In most developed nations, the sodium content of the adult diet is high, and salt craving in adults with 21-OHD is uncommon. In fact, children [29] and adults [30] with 21-OHD are prone to develop hypertension, prompting reduction or rarely discontinuation of mineralocorticoid dosing, or the paradoxical need for antihypertensive drugs plus fludrocortisone acetate. To titrate mineralocorticoid replacement, the serum potassium concentrations, plasma renin activity, and blood pressure in the sitting and standing positions are monitored, with the goals of renin as low as possible, normokalemia, and lack of orthostasis.
Glucocorticoid replacement therapy remains of central importance in the adult with 21-OHD, despite the disturbing fact that many patients will discontinue or neglect their treatment for months at a time. Hydrocortisone remains the safest drug for long-term replacement, as is the case for other forms of adrenal insufficiency. Regimens that require 3 doses a day, as is often used in children to suppress adrenal 19-carbon steroid production, pose compliance difficulties for some adults with busy schedules. Most patients manage to take 2 doses a day consistently, patterned after doses used for adrenal insufficiency, such as 15–20 mg on arising and 5–10 mg after the midday meal. The disadvantage of such a two-dose regimen is the tendency for ACTH to rise robustly in the early morning hours prior to the first dose, which worsens control, at least for a few hours until the first dose. In children, several groups have found empirically that administration of the largest dose at bedtime is associated with tightest control of adrenal steroidogenesis. Other groups have argued that the half-life of hydrocortisone is so short that an evening dose is only effective in suppressing the morning ACTH rise if a highly supraphysiologic dose is administered, such that hydrocortisone should be given during the day only [31]. Addition of an evening dose can cause poor sleep and worsen weight gain and other glucocorticoid-related side effects, but not in all patients. Unfortunately, well-designed and controlled studies comparing various regimens are lacking, so the clinician is advised to generally try the safest and most physiologic replacement regimen, and then to intensify therapy gradually if symptoms and signs of undertreatment become evident. Sustained-release hydrocortisone preparations administered once daily with promising pharmacokinetics have been developed [32], but these products currently are not widely available.
To simplify dosing and to increase suppression of ACTH, synthetic glucocorticoids such as prednisone, prednisolone, methylprednisolone, and dexamethasone may be employed. The problem with these drugs is that long-term usage almost invariably results in iatrogenic Cushing syndrome, due to their potency and long half-life. Dexamethasone needs only to be given once daily, with bedtime administration being the most effective means to suppress ACTH and adrenal 19-carbon steroid production [31]. In fact, the suppressive effect of single dose of dexamethasone on DHEAS production often lasts longer than 24 hours. Prednisolone or prednisone may also be used once daily as in adrenal insufficiency [33], but divided doses may be better in certain situations discussed below. Combined regimens, using more potent steroids only a few days per week, are more complicated, but this approach features the advantage of mitigating many of the side effects of taking these drugs chronically while utilizing their capacity to suppress DHEAS production longer and more completely than hydrocortisone.
Chronic ACTH excess juxtaposed with chronic glucocorticoid therapy, often at supraphysiologic doses, might place patients with 21-OHD at high risk for long-term complications [5]. The adrenal glands become progressively hyperplastic in most patients with 21-OHD, and adrenal tumors, including massive myelolipomas [34], have been described. In addition to these tumors, a paradoxically increased prevalence of hypertension has been found in adolescents [29] and adults [30] with 21-OHD. Reduced bone mineral density has been found in 21-OHD in most studies [35, 36], but the degree is moderate, and frank osteoporosis is uncommon. Other reports note a higher prevalence of glucose intolerance, obesity, and dyslipidemia in adults with 21-OHD compared to controls [30], raising their risk for cardiovascular disease [37]. These health problems are similar to those maladies associated with poor quality of life in patients with adrenal insufficiency [38] or cured Cushing's disease [39] after long-term followup. Curiously, concomitant androgen excess might mitigate some of the catabolic consequences of chronic glucocorticoid exposure, and some benefit is suggested by the observation that the increased risks appear not large, and major health problems are not consistently seen.
Above all, the management of the patient with 21-OHD should recognize that these patients are vulnerable to all the other diseases that people without this condition suffer, and they require the same general healthcare as any adult. Women should receive cervical smears, breast exams, and/or mammograms at the standard ages, and men require screening for prostate disease. Men and women should have blood pressure and cholesterol screening, colon cancer screening, education to avoid smoking and to follow a healthy lifestyle, and care for acute illnesses. Glucocorticoid doses should be increased for surgery and for acute illness when volume depletion from high fever, vomiting, diarrhea, or poor oral intake occurs—but not for any minor illness or injury.
3.2. Special Considerations for Women
In women with 21-OHD, two additional key issues are: (1) whether androgen excess symptoms are currently a problem, and (2) if the patient trying to conceive a child now. At a minimum, all women with 21-OHD must receive replacement doses of hydrocortisone and fludrocortisone acetate, as would any other patient with adrenal insufficiency. Higher doses and more potent steroids are used to the extent demanded by the two issues above. A normal serum testosterone and absence of androgen excess symptoms indicate adequate glucocorticoid treatment. If not, measurement of 17-hydroxyprogesterone and androstenedione, preferably in the follicular phase, will help to assess the degree of control. Chronic normalization of 17-hydroxyprogesterone indicates overtreatment and should be avoided, as values well above normal are acceptable if androgen concentrations are controlled and the patient is clinically well. The exception would be during specific times when fertility is sought.
Androgen excess is always a problem for these women, but the degree and its clinical consequences vary tremendously, and there is no reason to expose the woman to higher doses of corticosteroids if no benefit will be derived. If control has been poor in childhood, the patient seems to be more vulnerable to androgen excess, possibly due to greater adrenal hyperplasia. In addition, women with 21-OHD often develop a secondary polycystic ovary syndrome (PCOS), such that androgens and 17-OHP also derive from the ovaries and persists even after adrenal suppression [40]. Consequently, menses may remain irregular even with good control of the adrenal axis. If pregnancy is not desired, oral contraceptive pills provide several benefits, including lowering ovarian androgen production, maintaining monthly menses, and raising sex hormone binding globulin (SHBG), which lowers free testosterone and mitigates some of the hyperandrogenism with lower doses of glucocorticoid therapy. Antiandrogen therapy is a logical approach for treatment of 21-OHD, but with the exception of multidrug regimens [41], has received little attention. Spironolactone, an effective and inexpensive agent used to treat other forms of hirsutism and PCOS, is difficult to use in 21-OHD because it is both androgen and mineralocorticoid receptor antagonist, which causes salt loss. With the exception of flutamide [41], androgen antagonists and 5α-reductase inhibitors (finasteride and dutasteride) have not been formally studied in women with 21-OHD, although some patients have experimented with regimens containing these drugs. Topical agents, such as eflornithine cream, and mechanical methods (laser, electrolysis, shaving, and plucking) should not be neglected.
For women who are trying to become pregnant, 21-OHD introduces multiple barriers to conception. In the first place, a minority of women with 21-OHD even attempt to become pregnant [42], especially those with the most severe or "salt-wasting" disease [43]. The reasons for their not attempting pregnancy include vaginal abnormalities, such as inadequate caliber for coitus or strictures following repair [43] and dyspareunia [42]. Chronic hyperandrogenism adds physical and psychological challenges for these women. Finally, even in those who wish to become mothers, chronic oligoanovulation is common due to high androgens and secondary PCOS [40], whereas high circulating progesterone [44], which accumulates 2 steps behind the block, prevents favorable endometrial maturation and cervical mucus formation [45].
Unlike the multiple options available for women not attempting pregnancy, the only approach that will improve the hormonal milieu for ovulation and implantation is to intensify the glucocorticoid therapy, and the clinician should not be reluctant to use supraphysiologic dosing for several months to accomplish this purpose. In addition to controlling androgen excess, follicular phase progesterone should be suppressed below 2 nmol/L (0.6 ng/mL) to favor fertilization and implantation [46]. This goal can be achieved with prednisolone or hydrocortisone divided into 2 or more commonly 3 daily doses, as an evening dose is often necessary to lower morning serum progesterone adequately. A recent report demonstrated that 21/23 women with 21-OHD who desired pregnancy became pregnant and had live births using these regimens [46]. In these women, prenatal therapy is another issue if their spouse is a carrier; this topic is reviewed elsewhere.
Despite years of work and innovative surgical approaches, vaginal reconstruction remains a major problem in girls with 21-OHD. Often staged procedures are employed with the goal of urinary hygiene and appropriate clitoral reduction in the neonate. The trend now is to minimize surgery in the newborn and child and to leave complete establishment of an adequate vaginal orifice for when the woman becomes sexually active. Consequently, many adult women with 21-OHD face the need for definitive reconstruction surgery. The choice of procedure is important, but the woman must be motivated to use lubricants and dilatators regularly to obtain the best results and satisfaction. Traditional skin or bowel grafts often give suboptimal results, particularly in severely virilized girls [47]. Buccal mucosa grafts have shown good results in series with limited followup and may become a procedure of choice in the future [48, 49].
3.3. Special Considerations for Men
Men with 21-OHD are spared the disturbing and disfiguring effect of contrasexual hormone excess, which probably renders men with 21-OHD more prone to neglect their treatment than women. Unfortunately, men are not spared some of the long-term health consequences of both 21-OHD and glucocorticoid therapy, and male physiology and anatomy predispose these men to other problems. Adrenal androgen excess, when severe, can suppress gonadotropins and lead to testicular atrophy and infertility [50]. Reduced fertility has been observed in small cohorts of men with 21-OHD [51–53], but aberrant androgen metabolism is only one reason.
The second major cause of infertility and testicular dysfunction in men with 21-OHD is the presence and hyperplasia of adrenal rest tumors in the testes (TARTs). The prevalence of TART in men with 21-OHD is as high as 50%, if ascertained by sensitive ultrasonography or magnetic resonance imaging [51, 54]. These TARTs can become large and cause pain in addition to impaired testicular function. Treatment of the TARTs with intensified glucocorticoid replacement may cause regression and restoration of normal sperm counts [55].
At a minimum, men with 21-OHD should have an annual testicular exam, and screening ultrasonography is recommend by some authorities. If a mass is found, glucocorticoid treatment is intensified, such as dexamethasone 0.75–2 mg/d [55]. If the mass regresses, the treatment is continued for several weeks to months until a satisfactory result is obtained. If the mass does not shrink with dexamethasone, testicular cancer should be excluded by referral to an urologist. Surgical resection of the TARTs effectively controls testicular size and Leydig cell function but fails to restore fertility [56]. It is likely that, when a large TART is present, the testis is already damaged from increased pressure in the testicular capsule, which reduces testicular blood flow and obstructs the seminiferous tubules [52, 53]. Consequently, if preservation of fertility is a priority, the TARTs should be managed early and aggressively with medical therapy.
Replacement doses of hydrocortisone suffice for most men with 21-OHD, but which laboratory parameters should be monitored to guide glucocorticoid dosing is not known. If the 17-OHP is normal, the patient is overtreated, but values up to about 100 nmol/L ( 3,000 ng/dL) are probably a good target. The goal is to have a normal testosterone and normal gonadal function in the absence of TARTs, and men with a 17-OHP
3,000 ng/dL) are probably a good target. The goal is to have a normal testosterone and normal gonadal function in the absence of TARTs, and men with a 17-OHP  3,000 are unlikely to have large TARTs [57]. Measurement of adrostenedione and gonadotropins is appropriate if gonadal dysfunction becomes evident to determine if the adrenals or testes are the dominant source of androgens. The presence of TARTs prompts intensification of therapy to induce regression regardless of apparent hormonal control.
3,000 are unlikely to have large TARTs [57]. Measurement of adrostenedione and gonadotropins is appropriate if gonadal dysfunction becomes evident to determine if the adrenals or testes are the dominant source of androgens. The presence of TARTs prompts intensification of therapy to induce regression regardless of apparent hormonal control.
3.4. Indications for and Consequences of Bilateral Adrenalectomy Adults with 21-OHD
The curative potential of bilateral adrenalectomy for children with 21-OHD has been recognized, and its utility has been debated aggressively [58]. In adults with 21-OHD, unique indications have arisen. First, massive adrenal myelolipomas may develop [34] and can be bilateral [59] and require surgical removal. Second, although infertility in both men and women is often managed with intensified glucocorticoid therapy, adequate control cannot be achieved medically in some cases, and surgical management is required. Restoration of fertility in women [46, 60] and men [61] with 21-OHD after bilateral or unilateral adrenalectomy, respectively, has been observed and should be considered as an alternative approach.
4. Summary
The management of the adult with 21-OHD is a humbling and perilous process, given the lack of long-term data and comparative studies. Emerging evidence from centers with growing experience around the world is beginning to crystallize a set of guiding principles and concerns. The mere fact that we are now attentive to the long-term health consequences of this disease and its treatment attests to the success in the management of children with 21-OHD, which has been achieved over the last several decades. The challenges that remain are now to rene our approach to the adolescent and adult with 21-OHD, to develop better adjunctive treatments, and to optimize the quality of life for these patients. Therapy is expected to change over time as the norm rather than the exception. With each experience, we learn a little more about the physiology of adults with 21-OHD. Above all, the needs of the patient, their unique physiology, and their past experiences should be borne in mind when developing treatment regimens.
References
Speiser PW, White PC: Congenital adrenal hyperplasia. The New England Journal of Medicine. 2003, 349 (8): 776-788. 10.1056/NEJMra021561.
Therrell BL, Berenbaum SA, Manter-Kapanke V: Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998, 101 (4): 583-590. 10.1542/peds.101.4.583.
Wilkins L, Lewis RA, Klein R, Rosenberg E: The supression of androgen secretion by cortisone in a case of congenital adrenal hyperplasia. Bulletin of the Johns Hopkins Hospital. 1950, 86: 249-
Clayton PE, Oberfield SE, Martin Ritzén E: Consensus: consensus statement on 21-hydroxylase deficiency from The Lawson Wilkins Pediatric Endocrine Society and The European Society for Pediatric Endocrinology. Journal of Clinical Endocrinology and Metabolism. 2002, 87 (9): 4048-4053. 10.1210/jc.2002-020611.
Merke DP: Approach to the adult with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2008, 93 (3): 653-660.
Speiser PW, Agdere L, Ueshiba H, White PC, New MI: Aldosterone synthesis in salt-wasting congenital adrenal hyperplasia with complete absence of adrenal 21-hydroxylase. The New England Journal of Medicine. 1991, 324 (3): 145-149. 10.1056/NEJM199101173240302.
Nakamura Y, Hornsby PJ, Casson P: Type 5 17
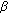 -hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (6): 2192-2198. 10.1210/jc.2008-2374.
-hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (6): 2192-2198. 10.1210/jc.2008-2374.Mapes S, Corbin CJ, Tarantal A, Conley A: The primate adrenal zona reticularis is defined by expression of cytochrome b5, 17
 -hydroxylase/17,20-lyase cytochrome P450 (P450c17) and NADPH-cytochrome P450 reductase (reductase) but not 3
-hydroxylase/17,20-lyase cytochrome P450 (P450c17) and NADPH-cytochrome P450 reductase (reductase) but not 3 -hydroxysteroid dehydrogenase/
-hydroxysteroid dehydrogenase/ isomerase (3
isomerase (3 -HSD). Journal of Clinical Endocrinology and Metabolism. 1999, 84 (9): 3382-3385. 10.1210/jc.84.9.3382.
-HSD). Journal of Clinical Endocrinology and Metabolism. 1999, 84 (9): 3382-3385. 10.1210/jc.84.9.3382.Nguyen AD, Mapes SM, Corbin CJ, Conley AJ: Morphological adrenarche in rhesus macaques: development of the zona reticularis is concurrent with fetal zone regression in the early neonatal period. Journal of Endocrinology. 2008, 199 (3): 367-378. 10.1677/JOE-08-0337.
Penning TM, Burczynski ME, Jez JM: Human 3
 -hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochemical Journal. 2000, 351 (1): 67-77. 10.1042/0264-6021:3510067.
-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochemical Journal. 2000, 351 (1): 67-77. 10.1042/0264-6021:3510067.Mahendroo MS, Wilson JD, Richardson JA, Auchus RJ: Steroid 5
 -reductase 1 promotes 5
-reductase 1 promotes 5 -androstane-3
-androstane-3 ,17
,17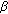 -diol synthesis in immature mouse testes by two pathways. Molecular and Cellular Endocrinology. 2004, 222: 113-120. 10.1016/j.mce.2004.04.009.
-diol synthesis in immature mouse testes by two pathways. Molecular and Cellular Endocrinology. 2004, 222: 113-120. 10.1016/j.mce.2004.04.009.Wilson JD, Auchus RJ, Leihy MW: 5
 -androstane-3
-androstane-3 ,17
,17 -diol is formed in tammar wallaby pouch young testes by a pathway involving 5
-diol is formed in tammar wallaby pouch young testes by a pathway involving 5 -pregnane-3
-pregnane-3 ,17
,17 -diol-20-one as a key intermediate. Endocrinology. 2003, 144: 575-580. 10.1210/en.2002-220721.
-diol-20-one as a key intermediate. Endocrinology. 2003, 144: 575-580. 10.1210/en.2002-220721.Biswas MG, Russell DW: Expression cloning and characterization of oxidative 17
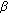 - and 3
- and 3 - hydroxysteroid dehydrogenases from rat and human prostate. The Journal of Biological Chemistry. 1997, 272 (25): 15959-15966. 10.1074/jbc.272.25.15959.
- hydroxysteroid dehydrogenases from rat and human prostate. The Journal of Biological Chemistry. 1997, 272 (25): 15959-15966. 10.1074/jbc.272.25.15959.Auchus RJ: The backdoor pathway to dihydrotestosterone. Trends in Endocrinology and Metabolism. 2004, 15 (9): 432-438.
Arlt W, Walker EA, Draper N: Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. The Lancet. 2004, 363 (9427): 2128-2135. 10.1016/S0140-6736(04)16503-3.
Shackleton C, Marcos J, Arlt W, Hauffa BP: Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): a disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype. American Journal of Medical Genetics. 2004, 129 (2): 105-112.
Shackleton C, Marcos J, Malunowicz EM: Biochemical diagnosis of Antley-Bixler syndrome by steroid analysis. American Journal of Medical Genetics. 2004, 128 (3): 223-231.
Homma K, Hasegawa T, Nagai T: Urine steroid hormone profile analysis in cytochrome P450 oxidoreductase deficiency: implication for the backdoor pathway to dihydrotestosterone. Journal of Clinical Endocrinology and Metabolism. 2006, 91 (7): 2643-2649. 10.1210/jc.2005-2460.
Ghayee H, Auchus R: Clinical implications of androgen synthesis via 5
 -reduced precursors. Endocrine Development. 2008, 13: 55-66.
-reduced precursors. Endocrine Development. 2008, 13: 55-66.Dalla Valle L, Toffolo V, Nardi A: The expression of the human steroid sulfatase-encoding gene is driven by alternative first exons. Journal of Steroid Biochemistry and Molecular Biology. 2007, 107 (1-2): 22-29. 10.1016/j.jsbmb.2007.05.004.
Lorence MC, Murry BA, Trant JM, Mason JI: Human 3
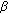 -hydroxysteroid dehydrogenase/
-hydroxysteroid dehydrogenase/ isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology. 1990, 126: 2493-2498. 10.1210/endo-126-5-2493.
isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology. 1990, 126: 2493-2498. 10.1210/endo-126-5-2493.Huang Y-W, Pineau I, Chang H-J: Critical residues for the specificity of cofactors and substrates in human estrogenic 17
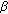 -hydroxysteroid dehydrogenase 1: variants designed from the three-dimensional structure of the enzyme. Molecular Endocrinology. 2001, 15 (11): 2010-2020. 10.1210/me.15.11.2010.
-hydroxysteroid dehydrogenase 1: variants designed from the three-dimensional structure of the enzyme. Molecular Endocrinology. 2001, 15 (11): 2010-2020. 10.1210/me.15.11.2010.Wu L, Einstein M, Geissler WM: Expression cloning and characterization of human 17
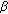 -hydrosteroid dehydrogenase type 2, a microsomal enzyme possessing 20
-hydrosteroid dehydrogenase type 2, a microsomal enzyme possessing 20 -hydroxysteroid dehydrogenase activity. The Journal of Biological Chemistry. 1993, 268: 12964-12969.
-hydroxysteroid dehydrogenase activity. The Journal of Biological Chemistry. 1993, 268: 12964-12969.Khan N, Sharma KK, Andersson S, Auchus RJ: Human 17-hydroxysteroid dehydrogenases types 1, 2, and 3 catalyze bi-directional equilibrium reactions, rather than unidirectional metabolism, in HEK-293 cells. Archives of Biochemistry and Biophysics. 2004, 429 (1): 50-59. 10.1016/j.abb.2004.05.023.
Jenkins EP, Andersson S, Imperato-McGinley J, Wilson JD, Russell DW: Genetic and pharmacological evidence for more than one human steroid 5
 -reductase. Journal of Clinical Investigation. 1992, 89 (1): 293-300. 10.1172/JCI115574.
-reductase. Journal of Clinical Investigation. 1992, 89 (1): 293-300. 10.1172/JCI115574.Turgeon D, Carrier JS, Levesque E, Hum DW, Belanger A: Relative enzymatic activity, protein stability, and tissue distribution of human steroid-metabolizing UGT2B subfamily members. Endocrinology. 2001, 142 (2): 778-787. 10.1210/en.142.2.778.
Winkel CA, Casey ML, Worley RJ, Madden JD, MacDonald PC: Extraadrenal steroid 21-hydroxylase activity in a woman with congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1983, 56 (1): 104-107. 10.1210/jcem-56-1-104.
Gomes LG, Huang N, Agrawal V: Extraadrenal 21-hydroxylation by CYP2C19 and CYP3A4: effect on 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (1): 89-95.
Nebesio TD, Eugster EA: Observation of hypertension in children with 21-hydroxylase deficiency: a preliminary report. Endocrine. 2006, 30 (3): 279-282. 10.1007/s12020-006-0005-4.
Falhammar H, Filipsson H, Holmdahl G: Metabolic profile and body composition in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (1): 110-116.
Charmandari E, Matthews DR, Johnston A, Brook CGD, Hindmarsh PC: Serum cortisol and 17-hydroxyprogesterone interrelation in classic 21-hydroxylase deficiency: is current replacement therapy satisfactory?. Journal of Clinical Endocrinology and Metabolism. 2001, 86 (10): 4679-4685. 10.1210/jc.86.10.4679.
Debono M, Ghobadi C, Rostami-Hodjegan A: Modified-release hydrocortisone to provide circadian cortisol profiles. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (5): 1548-1554. 10.1210/jc.2008-2380.
Caldato MC, Fernandes VT, Kater CE: One-year clinical evaluation of single morning dose prednisolone therapy for 21-hydroxylase deficiency. Arquivos Brasileiros de Endocrinologia e Metabologia. 2004, 48 (5): 705-712. 10.1590/S0004-27302004000500017.
Ravichandran R, Lafferty F, McGinniss MJ, Taylor HC: Congenital adrenal hyperplasia presenting as massive adrenal incidentalomas in the sixth decade of life: report of two patients with 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1996, 81 (5): 1776-1779. 10.1210/jc.81.5.1776.
Falhammar H, Filipsson H, Holmdahl G: Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (12): 4643-4649. 10.1210/jc.2007-0744.
Sciannamblo M, Russo G, Cuccato D, Chiumello G, Mora S: Reduced bone mineral density and increased bone metabolism rate in young adult patients with 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2006, 91 (11): 4453-4458. 10.1210/jc.2005-2823.
Kim MS, Merke DP: Cardiovascular disease risk in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Seminars in Reproductive Medicine. 2009, 27 (4): 316-321. 10.1055/s-0029-1225259.
Løvås K, Loge JH, Husebye ES: Subjective health status in Norwegian patients with Addison's disease. Clinical Endocrinology. 2002, 56 (5): 581-588. 10.1046/j.1365-2265.2002.01466.x.
Lindsay JR, Nansel T, Baid S, Gumowski J, Nieman LK: Long-term impaired quality of life in Cushing's syndrome despite initial improvement after surgical remission. Journal of Clinical Endocrinology and Metabolism. 2006, 91 (2): 447-453.
Barnes RB, Rosenfield RL, Ehrmann DA: Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. Journal of Clinical Endocrinology and Metabolism. 1994, 79 (5): 1328-1333. 10.1210/jc.79.5.1328.
Merke DP, Keil MF, Jones JV: Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2000, 85 (3): 1114-1120. 10.1210/jc.85.3.1114.
Gastaud F, Bouvattier C, Duranteau L: Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (4): 1391-1396. 10.1210/jc.2006-1757.
Nordenskjöld A, Holmdahl G, Frisen L: Type of mutation and surgical procedure affect long-term quality of life for women with congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2008, 93 (2): 380-386. 10.1210/jc.2007-0556.
Rosenfield RL, Bickel S, Razdan AK: Amenorrhea related to progestin excess in congenital adrenal hyperplasia. Obstetrics and Gynecology. 1980, 56 (2): 208-215.
Lo JC, Schwitzgebel VM, Tyrrell JB: Normal female infants born of mothers with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 1999, 84 (3): 930-936. 10.1210/jc.84.3.930.
Casteràs A, De Silva P, Rumsby G, Conway GS: Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clinical Endocrinology. 2009, 70 (6): 833-837. 10.1111/j.1365-2265.2009.03563.x.
Stikkelbroeck NM, Beerendonk CCM, Willemsen WNP: The long term outcome of feminizing genital surgery for congenital adrenal hyperplasia: anatomical, functional and cosmetic outcomes, psychosexual development, and satisfaction in adult female patients. Journal of Pediatric and Adolescent Gynecology. 2003, 16 (5): 289-296. 10.1016/S1083-3188(03)00155-4.
Lin WC, Chang CYY, Shen YY, Tsai HD: Use of autologous buccal mucosa for vaginoplasty: a study of eight cases. Human Reproduction. 2003, 18 (3): 604-607. 10.1093/humrep/deg095.
Samuelson ML, Baker LA: Autologous buccal mucosa vulvovaginoplasty for high urogenital sinus. Journal of Pediatric Urology. 2006, 2 (5): 486-488. 10.1016/j.jpurol.2005.09.001.
Tiitinen A, Välimäki M: Primary infertility in 45-year-old man with untreated 21-hydroxylase deficiency: successful outcome with glucocorticoid therapy. Journal of Clinical Endocrinology and Metabolism. 2002, 87 (6): 2442-2445. 10.1210/jc.87.6.2442.
Cabrera MS, Vogiatzi MG, New MI: Long term outcome in adult males with classic congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2001, 86 (7): 3070-3078. 10.1210/jc.86.7.3070.
Claahsen-van der Grinten HL, Otten BJ, Hermus AR, Sweep FC, Hulsbergen-van de Kaa CA: Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia can cause severe testicular damage. Fertility and Sterility. 2008, 89 (3): 597-601. 10.1016/j.fertnstert.2007.03.051.
Reisch N, Flade L, Scherr M: High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (5): 1665-1670. 10.1210/jc.2008-1414.
Avila NA, Premkumar A, Merke DP: Testicular adrenal rest tissue in congenital adrenal hyperplasia: comparison of MR imaging and sonographic findings. American Journal of Roentgenology. 1999, 172 (4): 1003-1006.
Claahsen-van der Grinten HL, Otten BJ, Sweep FC, Hermus AR: Repeated successful induction of fertility after replacing hydrocortisone with dexamethasone in a patient with congenital adrenal hyperplasia and testicular adrenal rest tumors. Fertility and Sterility. 2007, 88 (3): 705.e5-705.e8. 10.1016/j.fertnstert.2006.11.148.
Claahsen-van der Grinten HL, Otten BJ, Takahashi S: Testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia: evaluation of pituitary-gonadal function before and after successful testis-sparing surgery in eight patients. Journal of Clinical Endocrinology and Metabolism. 2007, 92 (2): 612-615.
Reisch N, Scherr M, Flade L: Total adrenal volume but not testicular adrenal rest tumor volume is associated with hormonal control in patients with 21-hydroxylase deficiency. Journal of Clinical Endocrinology and Metabolism. 2010, 95 (5): 2065-2072. 10.1210/jc.2009-1929.
Van Wyk JJ, Gunther DF, Ritzén EM: The use of adrenalectomy as a treatment for congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism. 1996, 81 (9): 3180-3190. 10.1210/jc.81.9.3180.
Sakaki M, Izaki H, Fukumori T: Bilateral adrenal myelolipoma associated with adrenogenital syndrome. International Journal of Urology. 2006, 13 (6): 801-802. 10.1111/j.1442-2042.2006.01406.x.
Ogilvie CM, Rumsby G, Kurzawinski T, Conway GS: Outcome of bilateral adrenalectomy in congenital adrenal hyperplasia: one unit's experience. European Journal of Endocrinology. 2006, 154 (3): 405-408. 10.1530/eje.1.02096.
Scaroni C, Favia G, Lumachi F: Unilateral adrenal tumor, erectile dysfunction and infertility in a patient with 21-hydroxylase deficiency: effects of glucocorticoid treatment and surgery. Experimental and Clinical Endocrinology and Diabetes. 2003, 111 (1): 41-43. 10.1055/s-2003-37499.
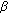 -hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (6): 2192-2198. 10.1210/jc.2008-2374.
-hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. Journal of Clinical Endocrinology and Metabolism. 2009, 94 (6): 2192-2198. 10.1210/jc.2008-2374. -hydroxylase/17,20-lyase cytochrome P450 (P450c17) and NADPH-cytochrome P450 reductase (reductase) but not 3
-hydroxylase/17,20-lyase cytochrome P450 (P450c17) and NADPH-cytochrome P450 reductase (reductase) but not 3 -hydroxysteroid dehydrogenase/
-hydroxysteroid dehydrogenase/ isomerase (3
isomerase (3 -HSD). Journal of Clinical Endocrinology and Metabolism. 1999, 84 (9): 3382-3385. 10.1210/jc.84.9.3382.
-HSD). Journal of Clinical Endocrinology and Metabolism. 1999, 84 (9): 3382-3385. 10.1210/jc.84.9.3382. -hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochemical Journal. 2000, 351 (1): 67-77. 10.1042/0264-6021:3510067.
-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochemical Journal. 2000, 351 (1): 67-77. 10.1042/0264-6021:3510067. -reductase 1 promotes 5
-reductase 1 promotes 5 -androstane-3
-androstane-3 ,17
,17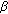 -diol synthesis in immature mouse testes by two pathways. Molecular and Cellular Endocrinology. 2004, 222: 113-120. 10.1016/j.mce.2004.04.009.
-diol synthesis in immature mouse testes by two pathways. Molecular and Cellular Endocrinology. 2004, 222: 113-120. 10.1016/j.mce.2004.04.009. -androstane-3
-androstane-3 ,17
,17 -diol is formed in tammar wallaby pouch young testes by a pathway involving 5
-diol is formed in tammar wallaby pouch young testes by a pathway involving 5 -pregnane-3
-pregnane-3 ,17
,17 -diol-20-one as a key intermediate. Endocrinology. 2003, 144: 575-580. 10.1210/en.2002-220721.
-diol-20-one as a key intermediate. Endocrinology. 2003, 144: 575-580. 10.1210/en.2002-220721.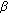 - and 3
- and 3 - hydroxysteroid dehydrogenases from rat and human prostate. The Journal of Biological Chemistry. 1997, 272 (25): 15959-15966. 10.1074/jbc.272.25.15959.
- hydroxysteroid dehydrogenases from rat and human prostate. The Journal of Biological Chemistry. 1997, 272 (25): 15959-15966. 10.1074/jbc.272.25.15959. -reduced precursors. Endocrine Development. 2008, 13: 55-66.
-reduced precursors. Endocrine Development. 2008, 13: 55-66.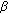 -hydroxysteroid dehydrogenase/
-hydroxysteroid dehydrogenase/ isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology. 1990, 126: 2493-2498. 10.1210/endo-126-5-2493.
isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology. 1990, 126: 2493-2498. 10.1210/endo-126-5-2493.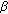 -hydroxysteroid dehydrogenase 1: variants designed from the three-dimensional structure of the enzyme. Molecular Endocrinology. 2001, 15 (11): 2010-2020. 10.1210/me.15.11.2010.
-hydroxysteroid dehydrogenase 1: variants designed from the three-dimensional structure of the enzyme. Molecular Endocrinology. 2001, 15 (11): 2010-2020. 10.1210/me.15.11.2010.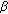 -hydrosteroid dehydrogenase type 2, a microsomal enzyme possessing 20
-hydrosteroid dehydrogenase type 2, a microsomal enzyme possessing 20 -hydroxysteroid dehydrogenase activity. The Journal of Biological Chemistry. 1993, 268: 12964-12969.
-hydroxysteroid dehydrogenase activity. The Journal of Biological Chemistry. 1993, 268: 12964-12969. -reductase. Journal of Clinical Investigation. 1992, 89 (1): 293-300. 10.1172/JCI115574.
-reductase. Journal of Clinical Investigation. 1992, 89 (1): 293-300. 10.1172/JCI115574.Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Auchus, R.J. Management of the Adult with Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol 2010, 614107 (2010). https://doi.org/10.1155/2010/614107
Received:
Accepted:
Published:
DOI: https://doi.org/10.1155/2010/614107