- Review Article
- Open access
- Published:
Growth and Reproductive Outcomes in Congenital Adrenal Hyperplasia
International Journal of Pediatric Endocrinology volume 2010, Article number: 298937 (2010)
Abstract
The treatment of congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency is complex. In addition to disease control, important therapeutic goals are the maintenance of normal growth and the acquisition of normal reproductive function. Here, data regarding final adult height (FH) in patients with CAH will be reviewed. Additional difficulties associated with CAH, including risks of obesity and hypertension, will be discussed. Information about fertility and reproductive outcomes in men and women with CAH will also be summarized. Although the treatment of each child with CAH needs to be individualized, close medical followup and laboratory monitoring along with good compliance can often result in positive clinical outcomes.
1. Final Adult Height in Congenital Adrenal Hyperplasia
The pediatric endocrinologist has the difficult task of carefully adjusting medications in actively growing children with CAH so as to avoid overtreatment as well as undertreatment. Glucocorticoid excess may result in poor linear growth, weight gain, hypertension, and other unwanted side effects. On the other hand, undertreatment results in excess androgen production and advanced skeletal maturation.
An important goal in treating children with CAH is to achieve a normal FH. Until recently, it was generally perceived that children with CAH would ultimately be short as adults and invariably below their genetic potential. However, studies over the last ten years have contrasted this idea (Table 1), suggesting that most children with CAH will have a FH that is below their target height but within 2 standard deviations (SD) of the mean.
Studies from 1977 through 1998 [1–17] describing height outcomes in patients with CAH were examined in a meta-analysis with respect to FH, impact of gender, and time of diagnosis [18]. Overall, mean weighted FH SD score (SDS) for all studies was  1.37. In regards to gender, mean FH SDS was
1.37. In regards to gender, mean FH SDS was  1.57 for males and
1.57 for males and  1.24 for females. When time at diagnosis was analyzed, mean FH was
1.24 for females. When time at diagnosis was analyzed, mean FH was  1.11 SDS for patients identified early (less than 1 year old) as compared to
1.11 SDS for patients identified early (less than 1 year old) as compared to  1.61 for patients indentified late (after 1 year old). Although not all reports are in agreement, more recent studies [18–33] also indicate that a FH within 2 SD of the mean can be achieved with males tending to have a slightly worse outcome than females (Table 1). Final adult height nearly equal to target height following strict compliance and regular clinic visits every three months has also been reported [34]. Therefore, there is now substantial evidence that with appropriate clinical management, most children with CAH can obtain a FH that is within their genetic potential [18, 35].
1.61 for patients indentified late (after 1 year old). Although not all reports are in agreement, more recent studies [18–33] also indicate that a FH within 2 SD of the mean can be achieved with males tending to have a slightly worse outcome than females (Table 1). Final adult height nearly equal to target height following strict compliance and regular clinic visits every three months has also been reported [34]. Therefore, there is now substantial evidence that with appropriate clinical management, most children with CAH can obtain a FH that is within their genetic potential [18, 35].
In growing children, it is well known that an elevated cumulative daily dose of glucocorticoids adversely affects linear growth. Glucocorticoids interfere with the normal interactions in the growth hormone (GH)/IGF-1 signaling cascade at the level of the hypothalamus, pituitary, and target organ as well as adversely affect hormone release, signal and gene transduction, and mRNA processing [36]. In human studies, glucocorticoid excess causes a decrease in GH response to growth hormone releasing hormone (GHRH) and a paradoxical increase in IGF-1 levels [37], creating a state of GH resistance. Although IGF-1 levels are elevated, studies in rats have shown that serum free IGF-1 levels are decreased after high-dose glucocorticoid administration [38]. In other animal studies, glucocorticoids decrease GH receptor synthesis and GH-induced IGF-1 mRNA expression [39]. Glucocorticoids also disturb normal calcium balance in the intestine and kidney and can interfere with osteoclast, osteoblast, and chondrocyte interaction [36]. In animal studies, glucocorticoids also induce apoptosis of chondrocytes through activation of caspases and inhibit the phosphatidylinositol  -kinase (PI3K) signaling pathway [40]. Thus, the negative impact of excess glucocorticoids on growth occurs through multiple different mechanisms. In general, hydrocortisone given three times a day has been associated with optimal outcomes [32]. Other forms of glucocorticoids, including prednisone and dexamethasone, have also been studied and are discussed in other sections of this supplement.
-kinase (PI3K) signaling pathway [40]. Thus, the negative impact of excess glucocorticoids on growth occurs through multiple different mechanisms. In general, hydrocortisone given three times a day has been associated with optimal outcomes [32]. Other forms of glucocorticoids, including prednisone and dexamethasone, have also been studied and are discussed in other sections of this supplement.
Higher doses of glucocorticoids in children with CAH result in decreased linear growth. In a prospective trial of 26 children with classic CAH, subjects were randomized to 15 mg/m2/day or 25 mg/ /day of hydrocortisone for six months and then crossed-over to the other dose [41]. Hydrocortisone was administered in three divided doses, and fludrocortisone was also given daily. Height velocity was significantly decreased during treatment with 25 mg/
/day of hydrocortisone for six months and then crossed-over to the other dose [41]. Hydrocortisone was administered in three divided doses, and fludrocortisone was also given daily. Height velocity was significantly decreased during treatment with 25 mg/ /day as compared with 15 mg/
/day as compared with 15 mg/ /day. A decrease in growth velocity was the most sensitive clinical indicator of glucocorticoid excess, and height velocity positively correlated with 17-hydroxyprogesterone concentrations [41]. The authors found that optimal growth occurred at an average plasma 17-hydroxyprogesterone concentration of 21 nmol/L (or 695 ng/dL). Thus, overzealous administration of glucocorticoids must be avoided.
/day. A decrease in growth velocity was the most sensitive clinical indicator of glucocorticoid excess, and height velocity positively correlated with 17-hydroxyprogesterone concentrations [41]. The authors found that optimal growth occurred at an average plasma 17-hydroxyprogesterone concentration of 21 nmol/L (or 695 ng/dL). Thus, overzealous administration of glucocorticoids must be avoided.
2. Growth during Critical Periods—Impact of Glucocorticoids and Androgen Exposure during Infancy and Puberty
Birth length in individuals with CAH was found in two reports to be significantly increased when compared to unaffected newborns [15, 42]. In one study [15], birth length of boys and girls diagnosed with CAH in infancy was statistically greater than the national mean birth lengths in Finland. In another study [42], the average birth length in boys (51.2  1.7 cm) and girls (50.3
1.7 cm) and girls (50.3  1.5 cm) with classic CAH was significantly greater than the mean birth length of unaffected Italian boys (50.7
1.5 cm) with classic CAH was significantly greater than the mean birth length of unaffected Italian boys (50.7  2.0 cm) and girls (49.4
2.0 cm) and girls (49.4  2.0 cm). Interestingly, the mean birth length in children with non-classic CAH did not differ significantly from the average birth length of Italian children [42]. These reports suggest that in utero hyperandrogenism can affect prenatal growth in severely affected children. Other studies have reported birth lengths in children with CAH that were mostly within the normal range [43].
2.0 cm). Interestingly, the mean birth length in children with non-classic CAH did not differ significantly from the average birth length of Italian children [42]. These reports suggest that in utero hyperandrogenism can affect prenatal growth in severely affected children. Other studies have reported birth lengths in children with CAH that were mostly within the normal range [43].
Infancy is a critically important time of growth and development. As is true in older children, impaired linear growth during infancy often reflects excess glucocorticoid therapy. In an Italian study [25], children with CAH who received a lower equivalent dose of hydrocortisone during the first year of life reached a better FH than those who received a higher dose. In particular, a negative influence on linear growth and ultimate height has been observed at hydrocortisone doses of more than 20 to 25 mg/ /day during infancy [14, 22, 44]. In a retrospective study from the Netherlands of 48 patients with salt-wasting CAH [45], a negative relationship between mean daily glucocorticoid dose during the first year of life and FH was noted.
/day during infancy [14, 22, 44]. In a retrospective study from the Netherlands of 48 patients with salt-wasting CAH [45], a negative relationship between mean daily glucocorticoid dose during the first year of life and FH was noted.
Interestingly, the negative effect of androgen excess on growth and skeletal maturation seen in later childhood does not occur during infancy. In a report from Sweden [46], children with a delayed diagnosis of CAH or who were undertreated early in life did not have growth acceleration or virilization until after 18 months of life, suggesting that growth prior to this time is insensitive to androgen exposure. In a study from the Netherlands [43], the growth pattern and skeletal maturation of 17 children with untreated simple-virilizing CAH revealed no increased height velocity in the first 12 months of life, which also suggests a relative resistance to androgen excess during infancy. However, after this time period, there was a progressive increase in growth velocity and bone maturation corresponding to the degree and duration of androgen exposure [43].
Another critically important time of growth in all children is early adolescence and puberty. In children with CAH, several studies have found impaired linear growth during puberty [19, 21–23, 32, 45]. After the age of 12 months and before 8 years, Stikkelbroeck et al. found no significant effect of glucocorticoid dose on height SDS [45]. However, between the ages of 8 and 14 years, there was a dose-dependent negative effect of glucocorticoids on linear growth. In other reports, the peak pubertal growth spurt occurred approximately 2 years earlier and the magnitude of the peak height velocity was lower in children with CAH when compared to controls [19, 47]. In a study comparing individuals treated with prednisone versus hydrocortisone, total pubertal growth was significantly decreased in both groups, and treatment with prednisone resulted in a decreased FH [32]. The dose at the start of puberty in the hydrocortisone-treated group was 16.7  4.1 mg/
4.1 mg/ /day versus 21.0
/day versus 21.0  8.1 mg/
8.1 mg/ /day in the prednisone-treated group using a relative glucocorticoid potency conversion of one for hydrocortisone and four for prednisone. The hydrocortisone equivalent dose at the onset of puberty correlated negatively with FH. In further analysis, the authors suggested that the deleterious effects on pubertal growth can be reduced if the hydrocortisone dose does not exceed 17 mg/
/day in the prednisone-treated group using a relative glucocorticoid potency conversion of one for hydrocortisone and four for prednisone. The hydrocortisone equivalent dose at the onset of puberty correlated negatively with FH. In further analysis, the authors suggested that the deleterious effects on pubertal growth can be reduced if the hydrocortisone dose does not exceed 17 mg/ /day [48]. Overtreatment of CAH at the onset of puberty may result in a less profound growth spurt due to an attenuated influence of sex steroids on growth [32]. Furthermore, inhibition of pituitary GH secretion during puberty may occur due to increased hypothalamic somatostatin release as a result of glucocorticoid excess [49, 50].
/day [48]. Overtreatment of CAH at the onset of puberty may result in a less profound growth spurt due to an attenuated influence of sex steroids on growth [32]. Furthermore, inhibition of pituitary GH secretion during puberty may occur due to increased hypothalamic somatostatin release as a result of glucocorticoid excess [49, 50].
Efforts to improve FH in children and adolescents with CAH have led researchers to investigate alternative and experimental adjunctive therapies, such as GH, gonadotropin-releasing hormone (GnRH) agonists, aromatase inhibitors, androgen blockers, or a combination of these medications. In a study from the United States [51], 12 children with CAH received GH and 8 received GH plus a GnRH analog. In both groups, growth rate and height prediction improved and there was a reduction in height deficit for bone age after one and two years of therapy [51, 52]. In a nonrandomized study of 14 patients with CAH who received both GH and a GnRH analog, FH and gain in height were statistically better when compared to a similar, untreated group [53]. Mean FH SDS was  0.4
0.4  0.8 in the treated group compared to
0.8 in the treated group compared to  1.4
1.4  1.1 in the untreated group. A different treatment strategy has used an antiandrogen (flutamide), an aromatase inhibitor (testolactone or letrozole), and a reduced hydrocortisone dose (less than 9 mg/m2/day) in children with CAH [54, 55]. In a randomized, cross-over short-term pilot study of 12 children using this experimental regimen compared to traditional hydrocortisone therapy, improved outcomes in linear growth, weight gain, and skeletal maturation were observed [54]. Two-year followup data were later published showing no adverse events and promising results despite elevated androgen levels in those on the experimental therapy [55]. Final height data and long-term safety results from this cohort of children have not yet been published.
1.1 in the untreated group. A different treatment strategy has used an antiandrogen (flutamide), an aromatase inhibitor (testolactone or letrozole), and a reduced hydrocortisone dose (less than 9 mg/m2/day) in children with CAH [54, 55]. In a randomized, cross-over short-term pilot study of 12 children using this experimental regimen compared to traditional hydrocortisone therapy, improved outcomes in linear growth, weight gain, and skeletal maturation were observed [54]. Two-year followup data were later published showing no adverse events and promising results despite elevated androgen levels in those on the experimental therapy [55]. Final height data and long-term safety results from this cohort of children have not yet been published.
3. Mineralocorticoid and Salt Replacement—Important Influences on Growth and Height Outcome
Besides glucocorticoids, mineralocorticoid and salt replacement in children with CAH also play an important role in linear growth. Mineralocorticoid therapy with fludrocortisone normalizes plasma renin activity and salt balance. Studies have shown that mineralocorticoid therapy in salt-wasting CAH improves linear growth and may allow for lower doses of glucocorticoids [56]. In a German study [34], a retrospective analysis of 39 adult patients with CAH found that a reduced daily dose of glucocorticoid was possible with the addition of daily fludrocortisone and was associated with improved FH. The authors proposed using a daily fludrocortisone dose of 110 mcg/m2 from preschool age onwards and suggested that this would result in a reduced mean hydrocortisone dose of approximately 13 mg/ /day in their patients. In a study from Italy [25], CAH patients treated early (before 3 years of age) with mineralocorticoid had an improved FH as compared to those treated late (after 5 years of age) or never. Final adult height in those treated early was 158.2
/day in their patients. In a study from Italy [25], CAH patients treated early (before 3 years of age) with mineralocorticoid had an improved FH as compared to those treated late (after 5 years of age) or never. Final adult height in those treated early was 158.2  5.6 cm (
5.6 cm ( 0.66
0.66  0.98 SDS) compared to 153.6
0.98 SDS) compared to 153.6  4.8 cm (
4.8 cm ( 1.49
1.49  0.79 SDS) in those treated late (
0.79 SDS) in those treated late ( ).
).
Salt supplementation during the first year of life has also been associated with improved linear growth and FH. In 34 patients from the Netherlands [23], half received supplemental salt at a dose of 2.5  3.0 mmol/kg/day (or approximately 150 mg/kg/day) from 0.19
3.0 mmol/kg/day (or approximately 150 mg/kg/day) from 0.19  0.15 years until 0.9
0.15 years until 0.9  0.38 years, and all received a similar dose of fludrocortisone. Mean length SDS at 2 years of life was higher in the salt supplemented group (
0.38 years, and all received a similar dose of fludrocortisone. Mean length SDS at 2 years of life was higher in the salt supplemented group ( 1.0 SDS) versus the group without salt supplementation (
1.0 SDS) versus the group without salt supplementation ( 1.56 SDS). Furthermore, FH corrected for target height was significantly higher in the salt supplemented group (
1.56 SDS). Furthermore, FH corrected for target height was significantly higher in the salt supplemented group ( 0.83 SDS) compared to the nonsupplemented group (
0.83 SDS) compared to the nonsupplemented group ( 1.69 SDS) (
1.69 SDS) ( ). The authors also found a positive correlation between FH SDS and salt supplementation during the first year of life.
). The authors also found a positive correlation between FH SDS and salt supplementation during the first year of life.
Complete aldosterone deficiency occurs in individuals with the most severe CYP21 gene mutations resulting in salt-wasting CAH. However, a study by Nimkarn et al. [57] has shown that there is some degree of aldosterone deficiency present in all forms of 21-hydroxylase deficiency, including simple-virilizing and non-classic forms. In an analysis of 402 individuals, the aldosterone to plasma renin activity ratio was abnormal in all forms of 21-hydroxylase deficiency, suggesting a spectrum of salt loss in CAH. Since salt-wasting crises do not occur in individuals with non-classic CAH, mineralocorticoid replacement is not required. Additional research is needed to see if mineralocorticoid replacement may benefit FH in all forms of CAH.
4. Other Complications—Weight Gain and Hypertension
Children with CAH are at an increased risk of developing obesity [58, 59]. Glucocorticoid dosage, chronological age, advanced skeletal maturation, and parental obesity were associated with an elevated body mass index (BMI) and obesity in a cross-sectional study of 89 children with CAH from Germany [59]. It is generally believed that excess glucocorticoid treatment in children with CAH results in weight gain and iatrogenic Cushing syndrome.
Another complication, which may or may not be related to weight gain or medications in the treatment of CAH, is hypertension [60]. Although hypertension is commonly discussed in the context of 11-beta-hydroxylase and 17-alpha-hydroxylase deficiencies due to elevated adrenal hormone metabolites with mineralocorticoid activity, it is not typically considered to be a component of 21-hydroxylase deficiency. Iatrogenic hypertension can occur from excess administration of medications with mineralocorticoid activity [61, 62]. Despite a reduction in the dose of fludrocortisone as well as a decrease in the dose of glucocorticoid, hypertension may persist in some individuals with CAH [62].
There are a few reports describing 24-hour ambulatory blood pressure monitoring in children and adolescents with CAH [63–66]. In a study by Roche et al. [63], 84% of children with salt-wasting CAH had absence of the normal physiologic nocturnal dip in systolic blood pressure. Blood pressure measurements were not correlated with any biochemical markers of CAH control. However, hypertension was strongly associated with obesity, particularly in females [63]. In another study, long-term glucocorticoid therapy did not correlate with blood pressure values [64]. In a report of 55 subjects with CAH in Germany [65], abnormal 24-hour blood pressure profiles and systolic hypertension correlated with the degree of overweight and obesity. Researchers found elevated serum leptin and insulin levels, which closely correlated to BMI and age, but laboratory markers of CAH control, glucocorticoid dose, and fludrocortisone dose did not correlate with blood pressure [65]. In another report of 23 children and adolescents with CAH, average blood pressure values were noted to be in the upper normal range [66].
As children with CAH are prone to increased weight gain and obesity, we examined our own population of CAH patients to see if an elevated BMI was a predictive factor in the development of hypertension [67]. Ninety-one patients with 21-hydroxylase deficiency were identified. Six (6.6%) were found to have hypertension. One child developed hypertension after presenting in an adrenal crisis with rhabdomyolysis and acute renal failure. Therefore, 5 children (5.5%) had essential hypertension, which was defined as hypertension of unknown etiology after an extensive evaluation by a pediatric cardiologist and pediatric nephrologist. Family history for hypertension was negative in all subjects who had essential hypertension. The age at diagnosis of hypertension ranged between 2 months to 12.6 years. All 5 children required anti-hypertensive medications, and only one child was able to eventually stop anti-hypertensive medication after 7 years of therapy. None of the subjects had a suppressed plasma renin activity at the time of diagnosis of hypertension, which implies that mineralocorticoid replacement was not excessive. The average dose of fludrocortisone at the time of diagnosis of hypertension was 0.09  0.05 mg/day. Also, the average dose of hydrocortisone at the time of diagnosis of hypertension was 16.4
0.05 mg/day. Also, the average dose of hydrocortisone at the time of diagnosis of hypertension was 16.4  1.6 mg/
1.6 mg/ /day, which suggests that excess glucocorticoid administration was not the culprit. Interestingly, an elevated BMI was not a determining factor in the development of hypertension. The average BMI was not statistically different between those without hypertension (23.9
/day, which suggests that excess glucocorticoid administration was not the culprit. Interestingly, an elevated BMI was not a determining factor in the development of hypertension. The average BMI was not statistically different between those without hypertension (23.9  9.1) as compared to those with essential hypertension (22.9
9.1) as compared to those with essential hypertension (22.9  2.9) [67]. The exact mechanism to explain why some individuals with CAH due to 21-hydroxylase deficiency develop hypertension is unclear, and further investigation is warranted.
2.9) [67]. The exact mechanism to explain why some individuals with CAH due to 21-hydroxylase deficiency develop hypertension is unclear, and further investigation is warranted.
5. Reproductive Outcome—Men
Compared to the general adult male population, men with CAH have reduced fertility [68]. While some males with CAH may develop hypogonadotropic hypogonadism due to high levels of sex steroids which suppress the hypothalamic-pituitary-gonadal axis, the most common reason for reduced fertility in men with CAH is the presence of testicular adrenal rest tumors (TARTs) [68, 69]. TARTs are usually not noticed by the patient or physician on physical exam unless they are greater than 2 cm [69]. Although most TARTs are nonpalpable, the majority of adolescent and adult males with CAH have one or more tumors. In a study of 17 adolescent and adult males with CAH [70], 6 individuals had palpable TARTs. In the remaining 11 patients, 10 had nonpalpable TARTs which were visible only by ultrasound. In the 11 patients who had semen analysis, abnormalities were noted in 7, and the best result was from the patient without any evidence of TART. In another study [20], 18 adult males with CAH had testicular ultrasounds and half were found to have TARTs. In the 9 affected patients, 7 had semen analysis and all were infertile. In a German study [71], 19 of 22 adult male patients with CAH were found to have an abnormal semen analysis and most were infertile.
TARTs are typically found in the rete testis, and more than 80% of patients have bilateral disease [69]. Due to their central location, TARTs can compress the seminiferous tubules and cause end-stage damage of the testicular parenchyma [72]. Testicular biopsies of 7 male patients with long-standing TARTs revealed decreased spermatogenesis and decreased tubular diameter with peritubular fibrosis [72]. There is no evidence that TARTs are malignant [68, 69]. However, long-standing TARTs can lead to irreversible testicular damage, and early recognition and treatment may prevent fertility problems [69, 72].
TARTs produce adrenal-specific steroids, contain adrenal-specific enzymes, and express ACTH and angiotensin II receptors [73]. Therefore, the first-line of treatment in a patient with a TART is to increase the glucocorticoid dose, which leads to suppression of ACTH secretion and may result in a decrease in size of the tumor and improvement in semen quality and fertility [74, 75]. In large, long-standing TARTs, increasing the dose of glucocorticoid may not be effective but removal of the tumor may prevent further testicular damage [69]. However, testis-sparing surgery may not improve gonadal function if permanent testicular damage has already occurred. In a study of 8 male CAH patients with bilateral TARTs and infertility [76], pituitary-gonadal testing and semen analysis was performed before and 6 and 22 months after testis-sparing surgery. Pituitary-gonadal function and semen analysis did not improve after surgery. Furthermore, all patients had persistently low inhibin B levels, indicating reduced Sertoli cell function. Although patients were followed for less than 2 years after surgery, these results suggest that testicular damage from TARTs may be irreversible if detected at a late stage.
Although most reports have investigated the prevalence of TARTs in adolescent and adult males with CAH, tumors have also been discovered in prepubertal males [77, 78]. In a study from the Netherlands [77], scrotal ultrasound was performed in 34 children with CAH between the ages of 2 and 18 years. TARTs were detected in 24% of children and most commonly in those with salt-wasting CAH. The youngest child noted to have TARTs was 7.25 years [77]. Markers of gonadal function, including LH, FSH, testosterone, and inhibin B levels, in children with and without TARTs was similar. In a different report from Chile [78], 19 children were studied between the ages of 2 and 10 years. TARTs were detected by ultrasound in 21%, and all had salt-wasting CAH. TARTs were detected in children aged 6.8, 7.3, 8.5, and 9.6 years. However, unlike the former study [77], markers of Sertoli and Leydig cell function were reduced, especially in those with elevated androgen levels indicating inadequate control of CAH [78].
Based upon the histological appearance and clinical observations of TARTs, including natural history and available treatment options, a five stage classification of TARTs (Table 2) has recently been proposed by a group from the Netherlands with extensive clinical experience [69, 79].
6. Reproductive Outcome—Women
When compared to unaffected women, adult women with CAH have decreased fertility rates [80–87]. The most widely recognized cause of low fertility rates in women with CAH is suboptimal disease control [88]. With adequate glucocorticoid and mineralocorticoid replacement, many sexually active women with CAH can become pregnant [89]. Even when the diagnosis of CAH is delayed and glucocorticoid replacement is not started until after age 9 years, chronic androgen exposure does not permanently or adversely affect fertility [90]. Other factors which may contribute to decreased fertility rates include ovarian hyperandrogenism and polycystic ovarian syndrome, complications related to genital surgery, and psychological factors [68].
In a review of pregnancy outcomes from 1956 through 2000 [82], 73 women with virilizing CAH had a total of 105 pregnancies. Women with the most severe, salt-wasting forms have the lowest fertility rates [82, 86]. Sexual function is reduced in women with CAH compared to controls, and it is lowest in women with the most severe degrees of virilization. In a study from France [85], 81% of women with CAH reported pain during vaginal penetration, and half were noted to have moderate or severe stenosis of their introitus. The live-birth rate in classic salt-wasting forms has been reported to be 0% to 10%, in simple virilizing forms 33% to 50%, and in non-classic forms 63% to 90% [84]. In a review by Lo and Grumbach [82], approximately 10% of pregnancies were spontaneously aborted and 10% had therapeutic abortions. Other reports have found a termination rate of approximately 6% [87]. The elective abortion rate of up to 10% may be due to women with CAH believing that their chance of becoming pregnant was impossible.
More recent studies have suggested improved pregnancy outcomes, which may be due to more stringent medical management with steroid replacement. In a report of 106 women with CAH from the United Kingdom [87], 21 of 23 trying to conceive achieved 34 pregnancies, which calculates to a pregnancy rate of 91.3% which is similar to the normal population (95%). However, the overall fertility rate was significantly lower than in the general population (0.25 versus 1.8). In those women who are able to conceive, pregnancy is generally uneventful [83, 91], although there is a higher incidence of delivery by cesarean section [91].
Unlike in males with CAH, adrenal rest tumors in the ovaries of females with CAH are extremely rare and therefore not a significant cause of decreased fertility [68]. There have been only 3 reports of females with CAH who were found to have ovarian adrenal rest tissue [92–94]. In a study of 13 adolescent and adult females with CAH, none were found to have ovarian adrenal rest tumors by ultrasound or magnetic resonance imaging [95].
Girls born to women with CAH are generally unaffected and have normal external genitalia since the placenta serves as a metabolic barrier to reduce fetal exposure to maternal androgens through placental aromatization of maternal testosterone and androstenedione [96]. There have been two reports of female infants born to mothers with CAH who had mild external virilization [97, 98]. In the first case, the infant was noted to have clitoral enlargement with normal vaginal and urethral openings and no labioscrotal fusion [97]. The other infant only had rugose labia majora with no clitoromegaly and a normal introitus [98]. To our knowledge, there has been only one reported case of a mother with classic, salt-wasting CAH having a female child with the same disorder [99]. Although not published, we are also aware of a male child at our institution with severe, salt-wasting CAH whose mother also had the same condition. Even though fertility is decreased, it is important to provide genetic counseling for adolescent and adult women with CAH. In addition, contraception counseling should also be considered with sexually active women with CAH since fertility is possible, especially in those women with good disease control.
7. Conclusion
With strict compliance to medical therapy as well as close clinical and laboratory monitoring, men and women with CAH can achieve a normal FH that is close to their calculated target height. Critical periods of growth when overtreatment with glucocorticoids should especially be avoided are during infancy and puberty. Replacement with salt and mineralocorticoid is also important in optimizing the linear growth of children with CAH. Side effects, which may or may not be related to glucocorticoid and mineralocorticoid replacement, include weight gain and hypertension. Fertility in men and women with CAH is reduced. In men, TARTs are the major cause of decreased fertility and early detection may help to reduce the risk of infertility. In women, fertility and reproductive outcome can be enhanced with improved biochemical control of CAH. Ongoing investigation of new therapies and novel strategies for clinical management will continue to enhance long-term growth and reproductive outcomes in individuals with CAH.
References
Klingensmith GJ, Garcia SC, Jones HW, Migeon CJ, Blizzard RM: Glucocorticoid treatment of girls with congenital adrenal hyperplasia: effects on height, sexual maturation, and fertility. The Journal of Pediatrics. 1977, 90 (6): 996-1004. 10.1016/S0022-3476(77)80581-7.
Ghali I, David M, David L: Linear growth and pubertal development in treated congenital adrenal hyperplasia due to 21 hydroxylase deficiency. Clinical Endocrinology. 1977, 6 (6): 425-436. 10.1111/j.1365-2265.1977.tb03325.x.
Urban MD, Lee PA, Migeon CJ: Adult height and fertility in men with congenital virilizing adrenal hyperplasia. The New England Journal of Medicine. 1978, 299 (25): 1392-1396. 10.1056/NEJM197812212992505.
Kirkland RT, Keenan BS, Holcombe JH, Kirkland JL, Clayton GW: The effect of therapy on mature height in congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 1978, 47 (6): 1320-1324. 10.1210/jcem-47-6-1320.
Clayton GW: Patterns of growth from birth to maturity in infants and children with congenital adrenal hyperplasia. Acta Endocrinologica. 1986, 113 (supplement 4): S295-S304.
DiMartino-Nardi J, Stoner E, O'Connell A, New MI: The effect of treatment of final height in classical congenital adrenal hyperplasia (CAH). Acta Endocrinologica. 1986, 113 (supplement 4): S305-S314.
Young MC, Ribeiro J, Hughes IA: Growth and body proportions in congenital adrenal hyperplasia. Archives of Disease in Childhood. 1989, 64 (11): 1554-1558. 10.1136/adc.64.11.1554.
Gargantini L, Calzi P, Brunelli V, Braggion F, Chiumello G: Growth and puberal development in males with adreno-genital syndrome. Pediatria Medica e Chirurgica. 1989, 11 (6): 597-602.
David M, Sempe M, Blanc M, Nicolino M, Forest MG, Morel Y: Final height in 69 patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Archives de Pediatrie. 1994, 1 (4): 363-367.
Kolousková S, Zemková D, Snajderová M, Lebl J: Growth and adult height in patients with congenital adrenal hyperplasia. Casopis Lekaru Ceskych. 1995, 134 (21): 689-691.
Lim YJ, Batch JA, Warne GL: Adrenal 21-hydroxylase deficiency in childhood: 25 years' experience. Journal of Paediatrics and Child Health. 1995, 31 (3): 222-227. 10.1111/j.1440-1754.1995.tb00790.x.
Yu ACM, Grant DB: Adult height in women with early-treated congenital adrenal hyperplasia (21-hydroxylase type): relation to body mass index in earlier childhood. Acta Paediatrica. 1995, 84 (8): 899-903. 10.1111/j.1651-2227.1995.tb13789.x.
Gussinyé M, Carrascosa A, Potau N: Bone mineral density in prepubertal and in adolescent and young adult patients with the salt-wasting form of congenital adrenal hyperplasia. Pediatrics. 1997, 100 (4): 671-674. 10.1542/peds.100.4.671.
Hauffa BP, Winter A, Stolecke H: Treatment and disease effects on short-term growth and adult height in children and adolescents with 21-hydroxylase deficiency. Klinische Padiatrie. 1997, 209 (2): 71-77. 10.1055/s-2008-1043931.
Jääskeläinen J, Voutilainen R: Growth of patients with 21-hydroxylase deficiency: an analysis of the factors influencing adult height. Pediatric Research. 1997, 41 (1): 30-33. 10.1203/00006450-199701000-00005.
Premawardhana LDKE, Hughes IA, Read GF, Scanlon MF: Longer term outcome in females with congenital adrenal hyperplasia (CAH): the Cardiff experience. Clinical Endocrinology. 1997, 46 (3): 327-332. 10.1046/j.1365-2265.1997.1360962.x.
Sellers E, Muirhead S, Guyda H: Adult height outcome in 21-hydroxylase deficiency congenital adrenal hyperplasia. Program and Abstracts, The Endocrine Society's 81st Annual Meeting. 1999, The Endocrine Society Press, 308-Poster P2-129, , Bethesda, Md, USA
Eugster EA, DiMeglio LA, Wright JC, Freidenberg GR, Seshadri R, Pescovitz OH: Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. The Journal of Pediatrics. 2001, 138 (1): 26-32. 10.1067/mpd.2001.110527.
Hargitai G, Sólyom J, Battelino T: Growth patterns and final height in congenital adrenal hyperplasia due to classical 21-hydroxylase deficiency results of a multicenter study. Hormone Research. 2001, 55 (4): 161-171. 10.1159/000049990.
Cabrera MS, Vogiatzi MG, New MI: Long term outcome in adult males with classic congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2001, 86 (7): 3070-3078. 10.1210/jc.86.7.3070.
Manoli I, Kanaka-Gantenbein Ch, Voutetakis A, Maniati-Christidi M, Dacou-Voutetakis C: Early growth, pubertal development, body mass index and final height of patients with congenital adrenal hyperplasia: factors influencing the outcome. Clinical Endocrinology. 2002, 57 (5): 669-676. 10.1046/j.1365-2265.2002.01645.x.
Muirhead S, Sellers EAC, Guyda H: Indicators of adult height outcome in classical 21-hydroxylase deficiency congenital adrenal hyperplasia. The Journal of Pediatrics. 2002, 141 (2): 247-252. 10.1067/mpd.2002.126601.
Van der Kamp HJ, Otten BJ, Buitenweg N: Longitudinal analysis of growth and puberty in 21-hydroxylase deficiency patients. Archives of Disease in Childhood. 2002, 87 (2): 139-144. 10.1136/adc.87.2.139.
Pinto G, Tardy V, Trivin C: Follow-up of 68 children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: relevance of genotype for management. The Journal of Clinical Endocrinology & Metabolism. 2003, 88 (6): 2624-2633. 10.1210/jc.2002-021433.
Balsamo A, Cicognani A, Baldazzi L: CYP21 genotype, adult height, and pubertal development in 55 patients treated for 21-hydroxylase deficiency. The Journal of Clinical Endocrinology & Metabolism. 2003, 88 (12): 5680-5688. 10.1210/jc.2003-030123.
Brunelli VL, Russo G, Bertelloni S: Final height in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: the Italian experience. Journal of Pediatric Endocrinology & Metabolism. 2003, 16 (supplement 2): 277-283.
Grigorescu-Sido A, Bettendorf M, Schulze E, Duncea I, Heinrich U: Growth analysis in patients with 21-hydroxylase deficiency influence of glucocorticoid dosage, age at diagnosis, phenotype and genotype on growth and height outcome. Hormone Research. 2003, 60 (2): 84-90. 10.1159/000071876.
Tung Y-C, Lee J-S, Tsai W-Y, Hsiao P-H: Adult height of patients with classical congenital adrenal hyperplasia. Journal of the Formosan Medical Association. 2005, 104 (2): 133-136.
Lemos-Marini SH, Guerra-Júnior G, Morcillo AM, Baptista MT, Silva LO, Maciel-Guerra AT: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: final height in 27 patients with the classical form. Arquivos Brasileiros de Endocrinologia e Metabologia. 2005, 49 (6): 902-907. 10.1590/S0004-27302005000600008.
Aycan Z, Ocal G, Berberoglu M, Cetinkaya E, Adiyaman P, Evliyaoglu O: Experience with long-term glucocorticoid treatment in congenital adrenal hyperplasia: growth pattern compared with genetic height potential. Journal of Pediatric Endocrinology & Metabolism. 2006, 19 (3): 245-251. 10.1515/JPEM.2006.19.3.245.
Sciannamblo M, Russo G, Cuccato D, Chiumello G, Mora S: Reduced bone mineral density and increased bone metabolism rate in young adult patients with 21-hydroxylase deficiency. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (11): 4453-4458. 10.1210/jc.2005-2823.
Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP: Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. The Journal of Clinical Endocrinology & Metabolism. 2007, 92 (5): 1635-1639. 10.1210/jc.2006-2109.
Chakhtoura Z, Bachelot A, Samara-Boustani D: Impact of total cumulative glucocorticoid dose on bone mineral density in patients with 21-hydroxylase deficiency. European Journal of Endocrinology. 2008, 158 (6): 879-887. 10.1530/EJE-07-0887.
Hoepffner W, Kaufhold A, Willgerodt H, Keller E: Patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency can achieve their target height: the Leipzig experience. Hormone Research. 2008, 70 (1): 42-50. 10.1159/000129677.
Dörr HG: Growth in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hormone Research. 2007, 68 (supplement 5): 93-99.
Hochberg Z: Mechanisms of steroid impairment of growth. Hormone Research. 2002, 58 (supplement 1): 33-38. 10.1159/000064764.
Borges M-HS, Pinto ACAR, DiNinno FB: IGF-I levels rise and GH responses to GHRH decrease during long-term prednisone treatment in man. Journal of Endocrinological Investigation. 1999, 22 (1): 12-17.
Skjærbæk C, Frystyk J, Grøfte T: Serum free insulin-like growth factor-I is dose-dependently decreased by methylprednisolone and related to body weight changes in rats. Growth Hormone & IGF Research. 1999, 9 (1): 74-80. 10.1054/ghir.1999.0090.
Beauloye V, Ketelslegers JM, Moreau B, Thissen JP: Dexamethasone inhibits both growth hormone (GH)-induction of insulin-like growth factor-I (IGF-I) mRNA and GH receptor (GHR) mrna levels in rat primary cultured hepatocytes. Growth Hormone & IGF Research. 1999, 9 (3): 205-211. 10.1054/ghir.1999.0110.
Chrysis D, Zaman F, Chagin AS, Takigawa M, Sävendahl L: Dexamethasone induces apoptosis in proliferative chondrocytes through activation of caspases and suppression of the Akt-phosphatidylinositol
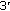 -kinase signaling pathway. Endocrinology. 2005, 146 (3): 1391-1397. 10.1210/en.2004-1152.
-kinase signaling pathway. Endocrinology. 2005, 146 (3): 1391-1397. 10.1210/en.2004-1152.Silva IN, Kater CE, Cunha CDF, Viana MB: Randomised controlled trial of growth effect of hydrocortisone in congenital adrenal hyperplasia. Archives of Disease in Childhood. 1997, 77 (3): 214-218. 10.1136/adc.77.3.214.
Balsamo A, Wasniewska M, Di Pasquale G: Birth length and weight in congenital adrenal hyperplasia according to the different phenotypes. European Journal of Pediatrics. 2006, 165 (6): 380-383. 10.1007/s00431-005-0075-y.
Claahsen-van der Grinten HL, Noordam K, Borm GF, Otten BJ: Absence of increased height velocity in the first year of life in untreated children with simple virilizing congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (4): 1205-1209. 10.1210/jc.2005-1701.
Einaudi S, Lala R, Corrias A, Matarazzo P, Pagliardini S, de Sanctis C: Auxological and biochemical parameters in assessing treatment of infants and toddlers with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The Journal of Pediatric Endocrinology. 1993, 6 (2): 173-178.
Stikkelbroeck NMML, Van't Hof-Grootenboer BAE, Hermus ARMM, Otten BJ, Van't Hof MA: Growth inhibition by glucocorticoid treatment in salt wasting 21-hydroxylase deficiency: in early infancy and (pre)puberty. The Journal of Clinical Endocrinology & Metabolism. 2003, 88 (8): 3525-3530. 10.1210/jc.2002-030011.
Thilen A, Woods KA, Perry LA, Savage MO, Wedell A, Ritzen EM: Early growth is not increased in untreated moderately severe 21-hydroxylase deficiency. Acta Paediatrica. 1995, 84 (8): 894-898. 10.1111/j.1651-2227.1995.tb13788.x.
Frisch H, Waldhauser F, Lebl J: Congenital adrenal hyperplasia: lessons from a multinational study. Hormone Research. 2002, 57 (supplement 2): 95-101. 10.1159/000058109.
Bonfig W, Pozza SBD, Schmidt H, Pagel P, Knorr D, Schwarz HP: Hydrocortisone dosing during puberty in patients with classical congenital adrenal hyperplasia: an evidence-based recommendation. The Journal of Clinical Endocrinology & Metabolism. 2009, 94 (10): 3882-3888. 10.1210/jc.2009-0942.
Lima L, Arce V, Diaz MJ, Tresguerres JAF, Devesa J: Glucocorticoids may inhibit growth hormone release by enhancing
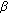 -adrenergic responsiveness in hypothalamic somatostatin neurons. The Journal of Clinical Endocrinology & Metabolism. 1993, 76 (2): 439-444. 10.1210/jc.76.2.439.
-adrenergic responsiveness in hypothalamic somatostatin neurons. The Journal of Clinical Endocrinology & Metabolism. 1993, 76 (2): 439-444. 10.1210/jc.76.2.439.Devesa J, Barros MG, Gondar M, Tresguerres JAF, Arce V: Regulation of hypothalamic somatostatin by glucocorticoids. Journal of Steroid Biochemistry and Molecular Biology. 1995, 53 (1–6): 277-282. 10.1016/0960-0760(95)00065-8.
Quintos JBQ, Vogiatzi MG, Harbison MD, New MI: Growth hormone therapy alone or in combination with gonadotropin-releasing hormone analog therapy to improve the height deficit in children with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2001, 86 (4): 1511-1517. 10.1210/jc.86.4.1511.
New MI: Factors determining final height in congenital adrenal hyperplasia. Journal of Pediatric Endocrinology & Metabolism. 2001, 14 (supplement 2): 933-937.
Lin-Su K, Vogiatzi MG, Marshall I: Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2005, 90 (6): 3318-3325. 10.1210/jc.2004-2128.
Laue L, Merke DP, Jones JV, Barnes KM, Hill S, Cutler GB: A preliminary study of flutamide, testolactone, and reduced hydrocortisone dose in the treatment of congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 1996, 81 (10): 3535-3539. 10.1210/jc.81.10.3535.
Merke DP, Keil MF, Jones JV, Fields J, Hill S, Cutler GB: Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2000, 85 (3): 1114-1120. 10.1210/jc.85.3.1114.
Kuhnle U, Rosler A, Pareira JA, Gunzcler P, Levine LS, New MI: The effects of long-term normalization of sodium balance on linear growth in disorders with aldosterone deficiency. Acta Endocrinologica. 1983, 102 (4): 577-582.
Nimkarn S, Lin-Su K, Berglind N, Wilson RC, New MI: Aldosterone-to-renin ratio as a marker for disease severity in 21-hydroxylase deficiency congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2007, 92 (1): 137-142. 10.1210/jc.2006-0964.
Cornean RE, Hindmarsh PC, Brook CGD: Obesity in 21-hydroxylase deficient patients. Archives of Disease in Childhood. 1998, 78 (3): 261-263. 10.1136/adc.78.3.261.
Völkl TMK, Simm D, Beier C, Dörr HG: Obesity among children and adolescents with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics. 2006, 117 (1): e98-e105. 10.1542/peds.2005-1005.
Nebesio TD: Hypertension in children with 21-hydroxylase deficiency. Congenital Adrenal Hyperplasia Research Education & Support (CARES) Foundation Newsletter. 2007, 6 (3): 14-15.
Vazquez AM, Kenny FM: Hypertension secondary to excessive desoxycorticosterone implants or 9-alpha fluorocortisol in salt-losing congenital adrenal hyperplasia. The Journal of Pediatrics. 1972, 81 (3): 549-552. 10.1016/S0022-3476(72)80188-4.
Kirkland JL, Kirkland RT, Librik L, Clayton GW: Iatrogenic hypertension in children with congenital adrenal hyperplasia. The Journal of Pediatrics. 1973, 83 (4): 687-689. 10.1016/S0022-3476(73)80243-4.
Roche EF, Charmandari E, Dattani MT, Hindmarsh PC: Blood pressure in children and adolescents with congenital adrenal hyperplasia (21-hydroxylase deficiency): a preliminary report. Clinical Endocrinology. 2003, 58 (5): 589-596. 10.1046/j.1365-2265.2003.01757.x.
de Silva KSH, Kanumakala S, Brown JJ, Jones CL, Warne GL: 24-hour ambulatory blood presurre profile in patients with congenital adrenal hyperplasia—a preliminary report. Journal of Pediatric Endocrinology & Metabolism. 2004, 17 (8): 1089-1095. 10.1515/JPEM.2004.17.8.1089.
Völkl TMK, Simm D, Dötsch J, Rascher W, Dörr HG: Altered 24-hour blood pressure profiles in children and adolescents with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (12): 4888-4895. 10.1210/jc.2006-1069.
Hoepffner W, Herrmann A, Willgerodt H, Keller E: Blood pressure in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Pediatric Endocrinology & Metabolism. 2006, 19 (5): 705-711. 10.1515/JPEM.2006.19.5.705.
Nebesio TD, Eugster EA: Observation of hypertension in children with 21-hydroxylase deficiency: a preliminary report. Endocrine. 2006, 30 (3): 279-282. 10.1007/s12020-006-0005-4.
Claahsen-van der Grinten HL, Stikkelbroeck NMML, Sweep CGJ, Hermus ARMM, Otten BJ: Fertility in patients with congenital adrenal hyperplasia. Journal of Pediatric Endocrinology & Metabolism. 2006, 19 (5): 677-685. 10.1515/JPEM.2006.19.5.677.
Claahsen-van der Grinten HL, Otten BJ, Stikkelbroeck MML, Sweep FCGJ, Hermus ARMM: Testicular adrenal rest tumours in congenital adrenal hyperplasia. Best Practice & Research: Clinical Endocrinology & Metabolism. 2009, 23 (2): 209-220. 10.1016/j.beem.2008.09.007.
Stikkelbroeck NMML, Otten BJ, Pasic A: High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2001, 86 (12): 5721-5728. 10.1210/jc.86.12.5721.
Reisch N, Flade L, Scherr M: High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2009, 94 (5): 1665-1670. 10.1210/jc.2008-1414.
Claahsen-van der Grinten HL, Otten BJ, Hermus ARMM, Sweep FCGJ, Hulsbergen-van de Kaa CA: Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia can cause severe testicular damage. Fertility and Sterility. 2008, 89 (3): 597-601. 10.1016/j.fertnstert.2007.03.051.
Claahsen-van der Grinten HL, Otten BJ, Sweep FCGJ: Testicular tumors in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency show functional features of adrenocortical tissue. The Journal of Clinical Endocrinology & Metabolism. 2007, 92 (9): 3674-3680. 10.1210/jc.2007-0337.
Claahsen-van der Grinten HL, Otten BJ, Sweep FCGJ, Hermus ARMM: Repeated successful induction of fertility after replacing hydrocortisone with dexamethasone in a patient with congenital adrenal hyperplasia and testicular adrenal rest tumors. Fertility and Sterility. 2007, 88 (3): 705.e5-705.e8. 10.1016/j.fertnstert.2006.11.148.
Mouritsen A, Juul A, Jørgensen N: Improvement of semen quality in an infertile man with 21-hydroxylase deficiency, suppressed serum gonadotropins and testicular adrenal rest tumours. International Journal of Andrology. 2009, 1-4.
Claahsen-van der Grinten HL, Otten BJ, Takahashi S: Testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia: evaluation of pituitary-gonadal function before and after successful testis-sparing surgery in eight patients. The Journal of Clinical Endocrinology & Metabolism. 2007, 92 (2): 612-615. 10.1210/jc.2006-1311.
Claahsen-van der Grinten HL, Sweep FCGJ, Blickman JG, Hermus ARMM, Otten BJ: Prevalence of testicular adrenal rest tumors in male children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. European Journal of Endocrinology. 2007, 157 (3): 339-344. 10.1530/EJE-07-0201.
Martinez-Aguayo A, Rocha A, Rojas N: Testicular adrenal rest tumors and Leydig and Sertoli cell function in boys with classical congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2007, 92 (12): 4583-4589. 10.1210/jc.2007-0383.
Claahsen-van der Grinten HL, Hermus ARMM, Otten BJ: Testicular adrenal rest tumours in congenital adrenal hyperplasia. International Journal of Pediatric Endocrinology. 2009, 2009: 8.
Mulaikal RM, Migeon CJ, Rock JA: Fertility rates in female patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The New England Journal of Medicine. 1987, 316 (4): 178-182. 10.1056/NEJM198701223160402.
Jääskeläinen J, Hippeläinen M, Kiekara O, Voutilainen R: Child rate, pregnancy outcome and ovarian function in females with classical 21-hydroxylase deficiency. Acta Obstetricia et Gynecologica Scandinavica. 2000, 79 (8): 687-692.
Lo JC, Grumbach MM: Pregnancy outcomes in women with congenital virilizing adrenal hyperplasia. Endocrinology and Metabolism Clinics of North America. 2001, 30 (1): 207-229. 10.1016/S0889-8529(08)70027-6.
Krone N, Wachter I, Stefanidou M, Roscher AA, Schwarz HP: Mothers with congenital adrenal hyperplasia and their children: outcome of pregnancy, birth and childhood. Clinical Endocrinology. 2001, 55 (4): 523-529. 10.1046/j.1365-2265.2001.01359.x.
Stikkelbroeck NMML, Hermus ARMM, Braat DDM, Otten BJ: Fertility in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Obstetrical & Gynecological Survey. 2003, 58 (4): 275-284.
Gastaud F, Bouvattier C, Duranteau L: Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2007, 92 (4): 1391-1396. 10.1210/jc.2006-1757.
Hagenfeldt K, Janson PO, Holmdahl G: Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Human Reproduction. 2008, 23 (7): 1607-1613. 10.1093/humrep/den118.
Conway GS, Casteràs A, De Silva P, Rumsby G: Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clinical Endocrinology. 2009, 70 (6): 833-837. 10.1111/j.1365-2265.2009.03563.x.
Meyer-Bahlburg HFL: What causes low rates of child-bearing in congenital adrenal hyperplasia?. The Journal of Clinical Endocrinology & Metabolism. 1999, 84 (6): 1844-1847. 10.1210/jc.84.6.1844.
Hoepffner W, Schulze E, Bennek J, Keller E, Willgerodt H: Pregnancies in patients with congenital adrenal hyperplasia with complete or almost complete impairment of 21-hydroxylase activity. Fertility and Sterility. 2004, 81 (5): 1314-1321. 10.1016/j.fertnstert.2003.10.024.
Kulshreshtha B, Marumudi E, Khurana ML: Fertility among women with classical congenital adrenal hyperplasia: report of seven cases where treatment was started after 9 years of age. Gynecological Endocrinology. 2008, 24 (5): 267-272. 10.1080/09513590801945230.
Dumic M, Janjanin N, Ille J: Pregnancy outcomes in women with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Journal of Pediatric Endocrinology & Metabolism. 2005, 18 (9): 887-895. 10.1515/JPEM.2005.18.9.887.
Russo G, Paesano P, Taccagni G, Del Maschio A, Chiumello G: Ovarian adrenal-like tissue in congenital adrenal hyperplasia. The New England Journal of Medicine. 1998, 339 (12): 853-854. 10.1056/NEJM199809173391220.
Al-Ahmadie HA, Stanek J, Liu J, Mangu PN, Niemann T, Young RH: Ovarian 'tumor' of the adrenogenital syndrome: the first reported case. American Journal of Surgical Pathology. 2001, 25 (11): 1443-1450. 10.1097/00000478-200111000-00015.
Claahsen-van der Grinten HL, Hulsbergen-van de Kaa CA, Otten BJ: Ovarian adrenal rest tissue in congenital adrenal hyperplasia—a patient report. Journal of Pediatric Endocrinology & Metabolism. 2006, 19 (2): 177-182. 10.1515/JPEM.2006.19.2.177.
Stikkelbroeck NMML, Hermus ARMM, Schouten D: Prevalence of ovarian adrenal rest tumours and polycystic ovaries in females with congenital adrenal hyperplasia: results of ultrasonography and MR imaging. European Radiology. 2004, 14 (10): 1802-1806. 10.1007/s00330-004-2329-x.
Lo JC, Schwitzgebel VM, Tyrrell JB: Normal female infants born of mothers with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The Journal of Clinical Endocrinology & Metabolism. 1999, 84 (3): 930-936. 10.1210/jc.84.3.930.
Kai H, Nose O, Iida Y, Ono J, Harada T, Yabuuchi H: Female pseudohermaphroditism caused by maternal congenital adrenal hyperplasia. The Journal of Pediatrics. 1979, 95 (3): 418-420. 10.1016/S0022-3476(79)80522-3.
Zacharin M: Fertility and its complications in a patient with salt losing congenital adrenal hyperplasia. Journal of Pediatric Endocrinology & Metabolism. 1999, 12 (1): 89-94. 10.1515/JPEM.1999.12.1.89.
Nebesio TD, Kreher NC, Hannon TS: Infant with classic congenital adrenal hyperplasia (CAH) born to a mother with classic CAH. The Journal of Pediatrics. 2004, 145 (2): 250-252. 10.1016/j.jpeds.2004.04.032.
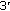 -kinase signaling pathway. Endocrinology. 2005, 146 (3): 1391-1397. 10.1210/en.2004-1152.
-kinase signaling pathway. Endocrinology. 2005, 146 (3): 1391-1397. 10.1210/en.2004-1152.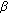 -adrenergic responsiveness in hypothalamic somatostatin neurons. The Journal of Clinical Endocrinology & Metabolism. 1993, 76 (2): 439-444. 10.1210/jc.76.2.439.
-adrenergic responsiveness in hypothalamic somatostatin neurons. The Journal of Clinical Endocrinology & Metabolism. 1993, 76 (2): 439-444. 10.1210/jc.76.2.439.Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nebesio, T.D., Eugster, E.A. Growth and Reproductive Outcomes in Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol 2010, 298937 (2010). https://doi.org/10.1155/2010/298937
Received:
Accepted:
Published:
DOI: https://doi.org/10.1155/2010/298937