- Review Article
- Open access
- Published:
46,XY DSD with Female or Ambiguous External Genitalia at Birth due to Androgen Insensitivity Syndrome, 5 -Reductase-2 Deficiency, or 17
-Reductase-2 Deficiency, or 17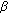 -Hydroxysteroid Dehydrogenase Deficiency: A Review of Quality of Life Outcomes
-Hydroxysteroid Dehydrogenase Deficiency: A Review of Quality of Life Outcomes
International Journal of Pediatric Endocrinology volume 2009, Article number: 567430 (2009)
Abstract
Disorders of sex development refer to a collection of congenital conditions in which atypical development of chromosomal, gonadal, or anatomic sex occurs. Studies of 46,XY DSD have focused largely on gender identity, gender role, and sexual orientation. Few studies have focused on other domains, such as physical and mental health, that may contribute to a person's quality of life. The current review focuses on information published since 1955 pertaining to psychological well-being, cognition, general health, fertility, and sexual function in people affected by androgen insensitivity syndromes, 5- reductase-2 deficiency, or 17
reductase-2 deficiency, or 17 -hydroxysteroid dehydrogenase-3 deficiency—reared male or female. The complete form of androgen insensitivity syndrome has been the focus of the largest number of investigations in domains other than gender. Despite this, all of the conditions included in the current review are under-studied. Realms identified for further study include psychological well-being, cognitive abilities, general health, fertility, and sexual function. Such investigations would not only improve the quality of life for those affected by DSD but may also provide information for improving physical and mental health in the general population.
-hydroxysteroid dehydrogenase-3 deficiency—reared male or female. The complete form of androgen insensitivity syndrome has been the focus of the largest number of investigations in domains other than gender. Despite this, all of the conditions included in the current review are under-studied. Realms identified for further study include psychological well-being, cognitive abilities, general health, fertility, and sexual function. Such investigations would not only improve the quality of life for those affected by DSD but may also provide information for improving physical and mental health in the general population.
1. Introduction
Disorders of sex development (DSD) refer to a collection of congenital conditions in which atypical development of sex occurs at one or more levels (chromosomal, gonadal, anatomic). Genetic males with DSD (i.e., 46,XY DSD) can present with an external genital phenotype that is female, ambiguous, or male including a micropenis (stretched penile length  2.5 SD for age). Studies of 46,XY DSD have focused largely on aspects of gender development such as satisfaction with gender of rearing, sexual orientation, gender identity, and gender role (GI/R) [1]. Very few studies have reported on additional aspects of physical or mental health despite the fact that patients and families often inquire about such topics. Furthermore, few studies have addressed the question of if, or how, a DSD diagnosis and related treatment impact quality of life (QoL) for affected individuals. For the purpose of this review, QoL can be operationally defined as the amount of enjoyment a person experiences in the physical, psychological, social, and spiritual dimensions of their life [2].
2.5 SD for age). Studies of 46,XY DSD have focused largely on aspects of gender development such as satisfaction with gender of rearing, sexual orientation, gender identity, and gender role (GI/R) [1]. Very few studies have reported on additional aspects of physical or mental health despite the fact that patients and families often inquire about such topics. Furthermore, few studies have addressed the question of if, or how, a DSD diagnosis and related treatment impact quality of life (QoL) for affected individuals. For the purpose of this review, QoL can be operationally defined as the amount of enjoyment a person experiences in the physical, psychological, social, and spiritual dimensions of their life [2].
Although much remains to be learned concerning psychosexual development as it relates to DSD, the purpose of this paper is to review what is known about aspects of physical and mental health that contribute to overall QoL beyond GI/R and sexual orientation. Topics reviewed include QoL and psychological well-being, cognition, general health, fertility, and sexual function. Additionally, the focus of this review is on individuals affected by 46,XY DSD due to androgen insensitivity syndromes (AIS), 5- reductase-2 deficiency (5
reductase-2 deficiency (5 -RD-2), or 17
-RD-2), or 17 -hydroxysteroid dehydrogenase-3 deficiency (17
-hydroxysteroid dehydrogenase-3 deficiency (17 -HSD-3)—reared male or female. These conditions were chosen because while much has been written about gender development in affected individuals, less emphasis has been placed on other outcomes that may contribute to QoL such as genital surgery or hormone therapy to induce and maintain pubertal development. Congenital micropenis was not included in the current review because too little information exists on outcomes aside from gender development in this DSD for meaningful interpretation of the data. Data on individuals with micropenis is further confused by the incorrect practice of some authors of describing individuals with a small phallus including hypospadias as having a micropenis.
-HSD-3)—reared male or female. These conditions were chosen because while much has been written about gender development in affected individuals, less emphasis has been placed on other outcomes that may contribute to QoL such as genital surgery or hormone therapy to induce and maintain pubertal development. Congenital micropenis was not included in the current review because too little information exists on outcomes aside from gender development in this DSD for meaningful interpretation of the data. Data on individuals with micropenis is further confused by the incorrect practice of some authors of describing individuals with a small phallus including hypospadias as having a micropenis.
2. Methods
A search of the databases Medline and Psyc INFO was performed using the terms disorders of sex development, DSD, intersex, hermaphrodite, male pseudohermaphrodite, ambiguous genitalia, androgen insensitivity, 5- reductase-2 deficiency, and 17
reductase-2 deficiency, and 17 -hydroxysteroid dehydrogenase-3 deficiency. Peer-reviewed articles published in English since 1955 were considered if they referred to adults with DSD grouped and analyzed according to the above-mentioned etiologies. The year 1955 was chosen because this was when John Money started to publish extensively on DSD in peer-reviewed scientific journals [3, 4].
-hydroxysteroid dehydrogenase-3 deficiency. Peer-reviewed articles published in English since 1955 were considered if they referred to adults with DSD grouped and analyzed according to the above-mentioned etiologies. The year 1955 was chosen because this was when John Money started to publish extensively on DSD in peer-reviewed scientific journals [3, 4].
To be considered for inclusion in the current review, articles must report on mental or physical health outcomes other than, or in addition to, GI/R and sexual orientation. Articles were excluded if they provided outcome information about GI/R or sexual orientation only, or when data from study participants could not be attributed to one of the specific DSD diagnoses referred to earlier. A total of 35 articles were located and reviewed according to these criteria.
3. Androgen Insensitivity Syndromes (AISs)
AISs in either the complete (CAIS) or partial (PAIS) form represent a relatively common presentation of 46,XY DSD [5–7]. CAIS and PAIS result in complete or partial end organ insensitivity to androgens, respectively. The majority of mutations of the human androgen receptor gene are base substitutions, although base and gene deletions, base insertions, and premature terminations occur. While it is well established that androgens bind to intracellular androgen receptors to alter gene expression in target tissues, evidence for nongenomic actions of androgens is accumulating [8]. The degree of androgen insensitivity of target tissues is usually inferred by the extent of under-masculinization of the external genitalia at birth coupled with the degree of under-virilization at puberty.
The clinical presentation of CAIS in adulthood is typically that of a tall woman with a female distribution of adipose tissue, female breasts and external genital development, and little or no sexual hair. Infants can present with testes that descend into the inguinal canals or labia, and adolescents can present with absent menses and sexual hair in conjunction with normal breast development. Due to the female external genital phenotype associated with CAIS, affected individuals are always assigned and reared as girls despite their possession of a 46,XY chromosomal complement and testes. Individuals with PAIS typically present as newborns with varying degrees of genital ambiguity. Thus, male or female sex of rearing occurs, depending on the degree of under-masculinization of the genitalia.
3.1. Health-Related Quality of Life, Psychological Well-Being, and Affective Disorders in CAIS
While psychosexual evaluations of girls and women affected by CAIS unequivocally reveal female GI/R [9–11], outcomes of other aspects of mental health vary greatly across studies. For example, psychological distress as assessed by the Brief Symptom Inventory (BSI), self-harming behavior, and suicidal tendencies are prevalent in some samples of women with CAIS recruited from physicians or support groups [12–14]. In a study of women with CAIS recruited from subspecialty clinics at a University Hospital, participants affected by CAIS reported better QoL scores and fewer depressive symptoms than women with other types of DSD or unaffected control women when assessed with the Danish version of the Quality of Life-Assessment of Growth Hormone Deficiency in Adults (QoL-AGHDA) questionnaire [15]. In yet another study of women with CAIS recruited from both a clinic sample and a support group, affected individuals did not differ from controls on measures of self-esteem, assessed by the Self-Esteem Scale, or psychological well-being, measured by responses to the Psychological General Well-Being Scale (PGWBS) [12]. While it is surprising that some studies of women with CAIS report equal or better QoL than unaffected women, this may be due to small sample sizes and participation bias. Additional QoL studies including larger sample sizes and greater participant diversity are needed in this population to better understand the impact of having a 46,XY chromosomal complement and being born with testes on overall psychological well-being in this group of women. Recently, the German Network of Disorders of Sex Development published clinical evaluation data on 439 individuals with DSD [16]. QoL information from this study sample will prove extremely valuable when published. Furthermore, a study that assesses potential differences in long-term QoL among women with CAIS whose testes were removed during infancy, compared to those who consented to gonadectomy later in their development, would provide very useful information for optimizing medical and surgical treatment for this group.
3.2. Cognition in CAIS
Mice with androgen receptor gene mutations (testicular feminization mutant or tfm) exhibit a more pronounced impairment on spatial recognition and memory retention tasks when considered in conjunction with the epsilon4 allele of the apolipoprotein E gene (apoE4) [17]. This subclass of apolipoprotein plays an important role in cholesterol metabolism and is associated with Alzheimer's disease, impaired cognition, and reduced neurite growth [18]. The implication is that androgen insensitivity may impact cognition in women affected by AIS who also express the epsilon 4 allele of the APOE gene.
Only two studies of cognitive performance in girls and women affected by CAIS exist in the peer-reviewed literature. The first employed standardized, age-appropriate intelligence testing for participants ranging in age from 5 to 28 years and observed normal scores for all of the girls and women who participated [19]. A second intelligence assessment was performed in women with CAIS and revealed no group differences in overall IQ between CAIS participants, unaffected men, and unaffected women [20]. However, unaffected women and those with CAIS performed worse than men on visuospatial components of the IQ measure. Taken together, these data reveal that girls and women with CAIS exhibit similar IQ scores and cognitive performance patterns as unaffected women who possess a 46,XX chromosomal complement and end organ responsiveness to androgens. Whether or not cognitive differences would be detected on measures of spatial recognition and memory for women affected by CAIS when apoE4 is considered is not known at this time. Future studies should consider the potential interaction of end organ unresponsiveness to androgens with apoE4 in this population. Additionally, more refined neuropsychological testing, regardless of apoE4 status, might reveal important cognitive differences in girls and women affected by this type of DSD.
3.3. General Health in CAIS
Few long-term outcome studies of general health have been conducted in women with CAIS. Obesity is commonly reported, however, at rates that mimic the general population of women [10, 21]. Decreased bone mineral densities in the lumbar spine and hip regions occur both prior to gonadectomy in women with CAIS as well as in gonadectomized women receiving daily estrogen therapy [10, 21, 22]. These data suggest that women affected by CAIS are at an increased risk for osteoporosis despite exposure to endogenous or exogenous estrogen. Whether women with CAIS require higher doses of estrogen than their unaffected counterparts to protect their bones, or whether androgens play a direct role in bone health for this group, is unknown at this time.
Concerning cancer occurrence and risk, both germ cell tumor formation and risk factors for developing prostate cancer have been investigated in women with CAIS. Prepubertal girls with CAIS are considered to be at low risk ( 2%) for germ cell tumor formation, as determined by review of 55 patients [23]. However, a seminoma has been reported in a 14-year-old with CAIS [24], and a malignant teratoma has been reported in a second affected child (also 14-years-old) [25]. In general, the risk for developing testicular tumors—including sertoli cell tumors, seminomas, and leydig cell tumors—is thought to increase with age in affected women [26, 27]. Additionally, women with CAIS appear to be at low risk for developing prostate cancer as determined by prostate specific antigen (PSA) testing and digital rectal exam, despite their possession of some prostate tissue [28]. No studies were located that investigated other types of cancer occurrence or risk in women with CAIS.
2%) for germ cell tumor formation, as determined by review of 55 patients [23]. However, a seminoma has been reported in a 14-year-old with CAIS [24], and a malignant teratoma has been reported in a second affected child (also 14-years-old) [25]. In general, the risk for developing testicular tumors—including sertoli cell tumors, seminomas, and leydig cell tumors—is thought to increase with age in affected women [26, 27]. Additionally, women with CAIS appear to be at low risk for developing prostate cancer as determined by prostate specific antigen (PSA) testing and digital rectal exam, despite their possession of some prostate tissue [28]. No studies were located that investigated other types of cancer occurrence or risk in women with CAIS.
3.4. Fertility and Sexual Function in CAIS
As women with CAIS possess neither ovaries nor a uterus, fertility is not possible in this group at the present time. While vaginal lengthening is needed for penile penetration in some women with CAIS [29, 30], others have a normal vaginal length and report satisfactory intercourse despite never having received dilatation or surgery [10, 31]. Three studies of orgasmic function report that women with CAIS can reach sexual climax [10, 32, 33], indicating that androgens are not necessary for this aspect of sexuality in this group. Low libido, or hyposexual function, is reported by some women with CAIS [33, 34]. Interestingly, even when women report sexual dysfunction, they also report sexual satisfaction at levels that equal unaffected women [34]. Perhaps hyposexual function associated with CAIS is secondary to vaginal hypoplasia, the need for androgens to support libido (but not orgasm), or sexual avoidance by women who feel stigmatized by their medical condition. Further research that includes unaffected women as control subjects is needed to understand the extent, and underlying causes, of hyposexual function in women affected by this 46,XY DSD. Additionally, future studies would benefit from considering the impact of the type and timing of vaginoplasty on sexual satisfaction—an important component of QoL.
3.5. Health-Related Quality of Life, Psychological Well-Being, and Affective Disorders in PAIS
Self-reported psychological distress as determined by responses to the BSI, self-harming behavior, suicidal tendencies, and suicidal attempts is observed in people affected by PAIS—whether reared male or female [12–14]. In a long-term followup study of adults with PAIS, psychological counseling for problems such as difficulty with family members, depression, and substance abuse was commonly reported during interviews—once again regardless of male or female rearing [35]. However, some studies of men and women affected by PAIS fail to observe significant problems with mental health [36, 37]. Furthermore, QoL as determined by responses to the SF-36 Health Survey exceeded normative data in one case series of 3 women affected by PAIS [38]. While it is clear that some individuals with PAIS experience mental health obstacles, both the extent and type of these obstacles in this patient population remain to be elucidated. The potential impact of clinical decisions such as the type and timing of genital surgeries on QoL in affected people, regardless of their gender assignment, is important information that is missing from our knowledge base at this time.
3.6. General Health and Cognition in PAIS
Based on studies of 24 patients, people with PAIS are thought to be at high risk ( 50%) for developing germ cell tumors if the testes are not located in the scrotum. For this group, gonadectomy is recommended at the time of diagnosis [23]. 46,XY women affected by PAIS do not appear to be at risk for developing prostate cancer, despite their possession of prostate tissue, as determined by PSA levels and digital rectal exam. Presumably this is due to the fact that these women are gonadectomized, and thus protected from androgenic actions on the prostate. In contrast, men with PAIS have PSA levels comparable to unaffected men matched for age and race [28]. Therefore, when individuals with PAIS are reared male, screening for prostate disease is recommended in a manner that is similar to the general male population. No other published reports on general health outcome in 46,XY individuals with PAIS, reared male or female, were located. Additionally, no studies on cognition in this group were found.
50%) for developing germ cell tumors if the testes are not located in the scrotum. For this group, gonadectomy is recommended at the time of diagnosis [23]. 46,XY women affected by PAIS do not appear to be at risk for developing prostate cancer, despite their possession of prostate tissue, as determined by PSA levels and digital rectal exam. Presumably this is due to the fact that these women are gonadectomized, and thus protected from androgenic actions on the prostate. In contrast, men with PAIS have PSA levels comparable to unaffected men matched for age and race [28]. Therefore, when individuals with PAIS are reared male, screening for prostate disease is recommended in a manner that is similar to the general male population. No other published reports on general health outcome in 46,XY individuals with PAIS, reared male or female, were located. Additionally, no studies on cognition in this group were found.
3.7. Fertility and Sexual Function in PAIS
At the present time fertility is challenging, but not impossible, for individuals with PAIS raised male [39, 40]. In contrast, fertility is not possible for individuals raised female. A study of 15 adults reared male found that none had ever engaged in penile-vaginal intercourse and all experienced severe sexual dysfunction [41]. In a second study of 21 men and 18 women with 46,XY DSD due to PAIS, most had participated in sexual relationships. Dissatisfaction with sexual function was common but not universal, and this dissatisfaction was similar for those reared male or female [35]. Smaller investigations of sexual function in women with PAIS reveal that, for those who participated in sexual relations with a partner, those experiences were rated as both satisfactory [37] and unsatisfactory [29, 37]. The great variability in outcome measures across studies indicates the need to study larger sample sizes [16] as well as employ standardized, validated measures of sexual function when examining men and women with PAIS.
4. 5 -Reductase-2 Deficiency
-Reductase-2 Deficiency
 -Reductase-2 Deficiency
-Reductase-2 Deficiency5 -reductase-2 (5
-reductase-2 (5 -RD-2) deficiency can result in female external genitalia (with a normal sized or enlarged clitoris) and male internal sex ducts in people with a 46,XY chromosomal complement. This occurs because the enzyme 5
-RD-2) deficiency can result in female external genitalia (with a normal sized or enlarged clitoris) and male internal sex ducts in people with a 46,XY chromosomal complement. This occurs because the enzyme 5 -RD-2 is needed to convert testosterone to dihydrotestosterone (DHT). DHT has a greater affinity for the androgen receptor than testosterone and is required to masculinize the external genitalia, but not the internal male sex ducts, during fetal development. Testosterone alone is sufficient to support somatic virilization postnatally [42, 43].
-RD-2 is needed to convert testosterone to dihydrotestosterone (DHT). DHT has a greater affinity for the androgen receptor than testosterone and is required to masculinize the external genitalia, but not the internal male sex ducts, during fetal development. Testosterone alone is sufficient to support somatic virilization postnatally [42, 43].
4.1. Health-Related Quality of Life, Psychological Well-Being, and General Health in 5 -RD-2 Deficiency
-RD-2 Deficiency
 -RD-2 Deficiency
-RD-2 DeficiencyClinical distress, determined by responses to the BSI and suicidal ideation, is observed in 46,XY women affected by 5 -RD-2 deficiency, although studies of larger sample sizes are needed to determine the generalizability of this finding [14]. Whether this distress is due to female assignment, feminizing surgeries, or both is unknown at this time. A bone health investigation of affected adults (none of whom had been gonadectomized nor given exogenous hormone replacement) revealed that bone mineral density did not differ from unaffected men [22]. A second bone density study concluded that DHT is not needed to maintain normal bone health [44]. No other studies on general health outcomes were located for review for this particular group of DSD. Of particular concern is the lack of knowledge pertaining to germ cell tumor [23] or prostate cancer [28] risk for this group. Additionally, no studies of cognitive performance were located.
-RD-2 deficiency, although studies of larger sample sizes are needed to determine the generalizability of this finding [14]. Whether this distress is due to female assignment, feminizing surgeries, or both is unknown at this time. A bone health investigation of affected adults (none of whom had been gonadectomized nor given exogenous hormone replacement) revealed that bone mineral density did not differ from unaffected men [22]. A second bone density study concluded that DHT is not needed to maintain normal bone health [44]. No other studies on general health outcomes were located for review for this particular group of DSD. Of particular concern is the lack of knowledge pertaining to germ cell tumor [23] or prostate cancer [28] risk for this group. Additionally, no studies of cognitive performance were located.
4.2. Fertility and Sexual Function in 5 -RD-2 Deficiency
-RD-2 Deficiency
 -RD-2 Deficiency
-RD-2 DeficiencyBoth sperm production and paternity have been documented in men affected by 5 -RD-2 deficiency [43, 45–48]. High-dose androgen therapy improves virilization, erectile response and ejaculatory volume in individuals reared male [45]. For those who identify as women, sexual activity is reported to be satisfactory following vaginal dilatation [44]. Similar to AIS, it is not clear how sexual function in people with 5
-RD-2 deficiency [43, 45–48]. High-dose androgen therapy improves virilization, erectile response and ejaculatory volume in individuals reared male [45]. For those who identify as women, sexual activity is reported to be satisfactory following vaginal dilatation [44]. Similar to AIS, it is not clear how sexual function in people with 5 -RD-2 deficiency, reared male or female, contributes to QoL.
-RD-2 deficiency, reared male or female, contributes to QoL.
5. 17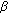 -Hydroxysteroid Dehydrogenase-3 Deficiency
-Hydroxysteroid Dehydrogenase-3 Deficiency
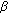 -Hydroxysteroid Dehydrogenase-3 Deficiency
-Hydroxysteroid Dehydrogenase-3 Deficiency17 -HSD-3 deficiency results in impaired testicular conversion of androstenedione to testosterone [49]. The clinical presentation of 17
-HSD-3 deficiency results in impaired testicular conversion of androstenedione to testosterone [49]. The clinical presentation of 17 -HSD-3 deficiency can be confused with CAIS as affected individuals often present with female external genitalia prior to puberty [50, 51]. If the testes remain in situ, virilization at puberty occurs in a manner that is similar to what is observed in 5
-HSD-3 deficiency can be confused with CAIS as affected individuals often present with female external genitalia prior to puberty [50, 51]. If the testes remain in situ, virilization at puberty occurs in a manner that is similar to what is observed in 5 -RD-2 deficiency [50, 52].
-RD-2 deficiency [50, 52].
5.1. Health-Related Quality of Life, Psychological Well-Being, and General Health in 17 -HSD-3 Deficiency
-HSD-3 Deficiency
 -HSD-3 Deficiency
-HSD-3 DeficiencyClinical distress as determined by the BSI and suicidal ideation is observed in 46,XY women affected by 17 -HSD-3 deficiency, but interpretation of these data is limited by the small sample size studied [14]. Once again, how gender assignment and the medical and surgical treatment that accompanies such assignment impact QoL for this group is unknown. Germ cell tumor risk is estimated to be intermediate (
-HSD-3 deficiency, but interpretation of these data is limited by the small sample size studied [14]. Once again, how gender assignment and the medical and surgical treatment that accompanies such assignment impact QoL for this group is unknown. Germ cell tumor risk is estimated to be intermediate ( 28%) [23, 50]. No other studies of general health or cognition conducted in this category of 46,XY DSD were located for review.
28%) [23, 50]. No other studies of general health or cognition conducted in this category of 46,XY DSD were located for review.
5.2. Sexual Function and Fertility in 17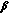 -HSD-3 Deficiency
-HSD-3 Deficiency
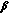 -HSD-3 Deficiency
-HSD-3 DeficiencyCase reports of 46,XY women with 17 -HSD-3 deficiency indicate satisfactory sexual function [44, 53], but available information is too incomplete to draw conclusions with confidence. Case reports of affected individuals living as men also indicate satisfactory sexual function [50, 53], although dissatisfaction attributed to having a small phallus appears in the literature [51].
-HSD-3 deficiency indicate satisfactory sexual function [44, 53], but available information is too incomplete to draw conclusions with confidence. Case reports of affected individuals living as men also indicate satisfactory sexual function [50, 53], although dissatisfaction attributed to having a small phallus appears in the literature [51].
6. Summary and Conclusions
Of the 35 studies reviewed that included outcomes information apart from gender, 24 included information about CAIS, 9 about PAIS, 11 about 5 -RD-2 deficiency, and 5 about 17
-RD-2 deficiency, and 5 about 17 -HSD-3 deficiency. Possibly, greater emphasis has been placed on studying CAIS apart from gender because female rearing in this particular DSD is undisputed. In contrast, male or female rearing occurs in people affected by PAIS, 5
-HSD-3 deficiency. Possibly, greater emphasis has been placed on studying CAIS apart from gender because female rearing in this particular DSD is undisputed. In contrast, male or female rearing occurs in people affected by PAIS, 5 -RD-2, and 17
-RD-2, and 17 -HSD-3 deficiencies. Perhaps GI/R and sexual orientation have been the focus of studies in these conditions in response to the clinical challenges of assigning gender in affected individuals. Finally, because CAIS has an animal model (tfm) associated with it, this condition may lend itself more easily to scientific investigation.
-HSD-3 deficiencies. Perhaps GI/R and sexual orientation have been the focus of studies in these conditions in response to the clinical challenges of assigning gender in affected individuals. Finally, because CAIS has an animal model (tfm) associated with it, this condition may lend itself more easily to scientific investigation.
Results from studies of psychological well-being in the specific 46,XY DSD categories considered here vary greatly across investigations. This is likely due to the fact that these include only a handful of participants, interviewed only once, in no particular relation to developmental stage or medical/surgical event. Future studies should include larger samples and take into account developmental stages as well as the potential impact of corresponding medical and surgical procedures associated with these stages [16]. Additionally, for those who have access to mental health services, it is critical to determine the effectiveness of these services on psychological well-being within specific categories of DSD.
No systematic studies of IQ or cognition have been conducted in people affected by 46,XY DSD due PAIS or androgen biosynthetic defects due to 5 -RD-2 or 17
-RD-2 or 17 -HSD-3 deficiencies. Unlike tfm rodent models, it is unknown if women with CAIS are more likely to develop spatial memory problems if they also express the epsilon 4 allele of the APOE gene. As individuals with 46,XY DSD are generally expected to live a full lifespan, a more complete understanding of the potential for their DSD to impact neuropsychological development, particularly later in development, is needed.
-HSD-3 deficiencies. Unlike tfm rodent models, it is unknown if women with CAIS are more likely to develop spatial memory problems if they also express the epsilon 4 allele of the APOE gene. As individuals with 46,XY DSD are generally expected to live a full lifespan, a more complete understanding of the potential for their DSD to impact neuropsychological development, particularly later in development, is needed.
General health outcome studies in people affected by AIS, 5 -RD-2 deficiency, and 17
-RD-2 deficiency, and 17 -HSD-3 deficiency are few and have been limited to gonadal tumors, and bone and prostate health. Further health outcome studies, particularly in conditions that exhibit clear sex differences or influences of sex steroid action, are needed. For example, whether or not possession of a Y chromosome places 46,XY women at risk for developing a male-typical presentation and course of cardiovascular disease, independent of androgen action, is not known. Alternatively, it is currently unknown if under-masculinized genetic males have an elevated risk for developing female-typical medical conditions such as autoimmune diseases or depression. As our understanding of DSD broadens to include physical and mental health outcomes apart from gender development, it is anticipated that QoL will improve for affected individuals.
-HSD-3 deficiency are few and have been limited to gonadal tumors, and bone and prostate health. Further health outcome studies, particularly in conditions that exhibit clear sex differences or influences of sex steroid action, are needed. For example, whether or not possession of a Y chromosome places 46,XY women at risk for developing a male-typical presentation and course of cardiovascular disease, independent of androgen action, is not known. Alternatively, it is currently unknown if under-masculinized genetic males have an elevated risk for developing female-typical medical conditions such as autoimmune diseases or depression. As our understanding of DSD broadens to include physical and mental health outcomes apart from gender development, it is anticipated that QoL will improve for affected individuals.
Fertility and sexual function can be important components to living a fulfilled life and maintaining relationships; yet we know very little about such topics. We do know from patient populations affected by conditions other than DSD that infertility [54] sexual dysfunction [55] and surgeries to the reproductive organs [56] negatively impact health-related QoL. Such information, as it pertains specifically to DSD, is crucial in optimizing treatment and supporting affected individuals.
Knowledge about the physical and mental health outcomes in DSD throughout the life cycle is important for improving or maintaining health in affected individuals and the population overall. For example, the few studies of bone health that have been conducted in AIS and 5 -RD-2 deficiency imply that testosterone, but not DHT, is necessary for optimizing bone density. Such information may be useful for developing treatments for conditions such as osteopenia and osteoporosis.
-RD-2 deficiency imply that testosterone, but not DHT, is necessary for optimizing bone density. Such information may be useful for developing treatments for conditions such as osteopenia and osteoporosis.
It is important to focus on QoL factors for persons with a DSD diagnosis because parents ask questions such as How will my child perform in school? Will my child establish friendships? Will my child have a career? Will my child fall in love? Parents also want to know how their children will feel about their DSD diagnosis. In short, parents want information about their affected child's future QoL. Only through systematic investigation of such questions will answers be obtained, and appropriate interventions developed.
We could find no data pertaining to individual adjustment to the categories of DSD discussed in the current review. Additionally, very little is known about how family members respond to a child's diagnosis of DSD. While we know that parents of children affected by CAIS or PAIS report feelings of shock, grief, anger, and shame when they learn of their child's condition [57], more studies of parents are needed as their attitudes surely exert a significant impact on subsequent child health and well-being [58]. Finally, the definition for QoL presented earlier includes social and spiritual components—neither of which were investigated systematically in any of the papers reviewed.
In conditions in which GI/R does not always develop in concordance with sex of rearing—such as PAIS, 5 -RD-2 deficiency, and 17
-RD-2 deficiency, and 17 -HSD-3 deficiency—a better understanding of factors that influence QoL may help to explain the developmental trajectory of GI/R in people for whom GI/R development does not match their initial gender assignment [11]. We still have a long way to go in understanding why some people with these conditions change gender while others do not. Importantly, approximately a third of people with DSD other than AIS, 5
-HSD-3 deficiency—a better understanding of factors that influence QoL may help to explain the developmental trajectory of GI/R in people for whom GI/R development does not match their initial gender assignment [11]. We still have a long way to go in understanding why some people with these conditions change gender while others do not. Importantly, approximately a third of people with DSD other than AIS, 5 -RD-2 deficiency, or 17
-RD-2 deficiency, or 17 -HSD-3 deficiency are born with malformations that likely affect QoL [59]. Future studies must consider those individuals as well. This review has attempted to show that we also have a long way to go before we have a full appreciation of the mental, physical, social, and spiritual domains that contribute to QoL of people affected by 46,XY DSD who are born with either female or ambiguous external genitalia.
-HSD-3 deficiency are born with malformations that likely affect QoL [59]. Future studies must consider those individuals as well. This review has attempted to show that we also have a long way to go before we have a full appreciation of the mental, physical, social, and spiritual domains that contribute to QoL of people affected by 46,XY DSD who are born with either female or ambiguous external genitalia.
References
Lee PA, Houk CP, Ahmed SF, Hughes IA: Consensus statement on management of intersex disorders. Pediatrics. 2006, 118 (2): e488-e500. 10.1542/peds.2006-0738.
Quality of Life in Health Promotion and Rehabilitation: Conceptual Approaches, Issues, and Applications. Edited by: Renwick R, Brown I, Nagler M. 1996, Sage, Thousand Oaks, Calif, USA
Money J, Hampson JG, Hampson JL: An examination of some basic sexual concepts: the evidence of human hermaphroditism. Bulletin of the Johns Hopkins Hospital. 1955, 97 (4): 301-319.
Money J, Hampson JG, Hampson JL: Hermaphroditismml: recommendations concerning assignment of sex, change of sex, and psychologic adjustment. Bulletin of the Johns Hopkins Hospital. 1955, 97 (4): 284-300.
Parisi MA, Ramsdell LA, Burns MW: A gender assessment teamml: experience with 250 patients over a period of 25 years. Genetics in Medicine. 2007, 9 (6): 348-357. 10.1097/GIM.0b013e3180653c47.
Al-Agha AE, Thomsett MJ, Batch JA: The child of uncertain sex: 17 years of experience. Journal of Paediatrics and Child Health. 2001, 37 (4): 348-351. 10.1046/j.1440-1754.2001.00669.x.
Ahmed SF, Khwaja O, Hughes IA: The role of a clinical score in the assessment of ambiguous genitalia. BJU International. 2000, 85 (1): 120-124. 10.1046/j.1464-410x.2000.00354.x.
Michels G, Hoppe UC: Rapid actions of androgens. Frontiers in Neuroendocrinology. 2008, 29 (2): 182-198. 10.1016/j.yfrne.2007.08.004.
Migeon CJ, Wisniewski AB, Brown TR: Androgen insensitivity syndrome. Hormone Resistance and Hypersensitivity States. Edited by: Chrousos GP. 2002, Lippincott Williams & Wilkins, Philadelphia, Pa, USA
Wisniewski AB, Migeon CJ, Meyer-Bahlburg HFL: Complete androgen insensitivity syndrome: long-term medical, surgical, and psychosexual outcome. The Journal of Clinical Endocrinology & Metabolism. 2000, 85 (8): 2664-2669. 10.1210/jc.85.8.2664.
Mazur T: Gender dysphoria and gender change in androgen insensitivity or micropenis. Archives of Sexual Behavior. 2005, 34 (4): 411-421. 10.1007/s10508-005-4341-x.
Hines M, Ahmed SF, Hughes IA: Psychological outcomes and gender-related development in complete androgen insensitivity syndrome. Archives of Sexual Behavior. 2003, 32 (2): 93-101. 10.1023/A:1022492106974.
Diamond M, Watson LA: Androgen insensitivity syndrome and Klinefelter's syndrome: sex and gender considerations. Child and Adolescent Psychiatric Clinics of North America. 2004, 13 (3): 623-640. 10.1016/j.chc.2004.02.015.
Schützmann K, Brinkmann L, Schacht M, Richter-Appelt H: Psychological distress, self-harming behavior, and suicidal tendencies in adults with disorders of sex development. Archives of Sexual Behavior. 2009, 38 (1): 16-33. 10.1007/s10508-007-9241-9.
Johannsen TH, Ripa CPL, Mortensen EL, Main KM: Quality of life in 70 women with disorders of sex development. European Journal of Endocrinology. 2006, 155 (6): 877-885. 10.1530/eje.1.02294.
Lux A, Kropf S, Kleinemeier E, Jürgensen M, Thyen U, The DSD Network Working Group : Clinical evaluation study of the German network of disorders of sex development (DSD)/intersexuality: study design, description of the study population, and data quality. BMC Public Health. 2009, 9, article 110:
Rizk-Jackson A, Robertson J, Raber J: Tfm-AR modulates the effects of ApoE4 on cognition. Journal of Neurochemistry. 2008, 105 (1): 63-67. 10.1111/j.1471-4159.2007.05092.x.
Bu G: Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nature Reviews Neuroscience. 2009, 10 (5): 333-344. 10.1038/nrn2620.
Masica DN, Money J, Ehrhardt AA, Lewis VG: IQ, fetal sex hormones and cognitive patterns: studies in the testicular feminizing syndrome of androgen insensitivity. The Johns Hopkins Medical Journal. 1969, 124 (1): 34-43.
Imperato-McGinley J, Pichardo M, Gautier T, Voyer D, Bryden MP: Cognitive abilities in androgen-insensitive subjects: comparison with control males and females from the same kindred. Clinical Endocrinology. 1991, 34 (5): 341-347. 10.1111/j.1365-2265.1991.tb00303.x.
Danilovic DLS, Correa PHS, Costa EMF, Melo KFS, Mendonca BB, Arnhold IJP: Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporosis International. 2007, 18 (3): 369-374. 10.1007/s00198-006-0243-6.
Sobel V, Schwartz B, Zhu Y-S, Cordero JJ, Imperato-McGinley J: Bone mineral density in the complete androgen insensitivity and
 -reductase-2 deficiency syndromes. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (8): 3017-3023. 10.1210/jc.2005-2809.
-reductase-2 deficiency syndromes. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (8): 3017-3023. 10.1210/jc.2005-2809.Looijenga LHJ, Hersmus R, Oosterhuis JW, Cools M, Drop SLS, Wolffenbuttel KP: Tumor risk in disorders of sex development (DSD). Best Practice and Research. 2007, 21 (3): 480-495. 10.1016/j.beem.2007.05.001.
Hurt WG, Bodurtha JN, McCall JB, Ali MM: Seminoma in pubertal patient with androgen insensitivity syndrome. American Journal of Obstetrics and Gynecology. 1989, 161 (3): 530-531.
Morris JM: The syndrome of testicular feminization in male pseudohermaphrodites. American Journal of Obstetrics and Gynecology. 1953, 65 (6): 1192-1211.
Iwamoto I, Yanazume S, Fujino T, Yoshioka T, Douchi T: Leydig cell tumor in an elderly patient with complete androgen insensitivity syndrome. Gynecologic Oncology. 2005, 96 (3): 870-872. 10.1016/j.ygyno.2004.11.015.
Nojima M, Taguchi T, Ando Y: Huge seminoma developed in a patient with testicular feminization. Journal of Obstetrics and Gynaecology Research. 2004, 30 (2): 109-112. 10.1111/j.1447-0756.2003.00168.x.
Salmasi AH, Wisniewski AB, Novak TE, Gearhart JP, Migeon CJ, Lakshmanan Y: Prostate screening in patients with 46,XY disorders of sex development—is it necessary?. The Journal of Urology. 2008, 180 (4): 1422-1426. 10.1016/j.juro.2008.06.043.
Costa EMF, Mendonca BB, Inácio M, Arnhold IJP, Silva FAQ, Lodovici O: Management of ambiguous genitalia in pseudohermaphrodites: new perspectives on vaginal dilation. Fertility and Sterility. 1997, 67 (2): 229-232. 10.1016/S0015-0282(97)81902-4.
Melo KFS, Mendonca BB, Billerbeck AEC: Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort: five novel mutations in the androgen receptor gene. The Journal of Clinical Endocrinology & Metabolism. 2003, 88 (7): 3241-3250. 10.1210/jc.2002-021658.
Purves JT, Miles-Thomas J, Migeon C, Gearhart JP: Complete androgen insensitivity: the role of the surgeon. The Journal of Urology. 2008, 180 (4, supplement 1): 1716-1719. 10.1016/j.juro.2008.03.110.
Morris JM, Mahesh VB: Further observations on the syndrome "testicular feminization". American Journal of Obstetrics & Gynecology. 1963, 87: 731-748.
Masica DN, Money J, Ehrhardt AA: Fetal feminization and female gender identity in the testicular feminizing syndrome of androgen insensitivity. Archives of Sexual Behavior. 1971, 1 (2): 131-142. 10.1007/BF01541057.
Minto CL, Liao KL-M, Conway GS, Creighton SM: Sexual function in women with complete androgen insensitivity syndrome. Fertility and Sterility. 2003, 80 (1): 157-164. 10.1016/S0015-0282(03)00501-6.
Migeon CJ, Wisniewski AB, Gearhart JP: Ambiguous genitalia with perineoscrotal hypospadias in 46,XY individuals: long-term medical, surgical, and psychosexual outcome. Pediatrics. 2002, 110 (3, article e31):
Money J, Ogunro C: Behavioral sexology: ten cases of genetic male intersexuality with impaired prenatal and pubertal androgenization. Archives of Sexual Behavior. 1974, 3 (3): 181-205. 10.1007/BF01541485.
Vates TS, Fleming P, Leleszi JP, Barthold JS, González R, Perlmutter AD: Functional, social and psychosexual adjustment after vaginal reconstruction. The Journal of Urology. 1999, 162 (1): 182-187. 10.1097/00005392-199907000-00065.
Mazur T, Sandberg DE, Perrin MA, Gallagher JA, MacGilivray MH: Male pseudohermaphroditismml: long-term quality of life outcome in five 46,XY individuals reared female. The Journal of Pediatric Endocrinology & Metabolism. 2004, 17 (6): 809-823. 10.1515/JPEM.2004.17.6.809.
Gooren L: Improvement of spermatogenesis after treatment with the antiestrogen tamoxifen in a man with the incomplete androgen insensitivity syndrome. The Journal of Clinical Endocrinology & Metabolism. 1989, 68 (6): 1207-1210. 10.1210/jcem-68-6-1207.
Giwercman A, Kledal T, Schwartz M: Preserved male fertility despite decreased androgen sensitivity caused by a mutation in the ligand-binding domain of the androgen receptor gene. The Journal of Clinical Endocrinology & Metabolism. 2000, 85 (6): 2253-2259. 10.1210/jc.85.6.2253.
Bouvattier C, Mignot B, Lefèvre H, Morel Y, Bougnères P: Impaired sexual activity in male adults with partial androgen insensitivity. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (9): 3310-3315. 10.1210/jc.2006-0218.
Wilson JD, Griffin JE, Russell DW: Steroid 5
 -reductase 2 deficiency. Endocrine Reviews. 1993, 14 (5): 577-593.
-reductase 2 deficiency. Endocrine Reviews. 1993, 14 (5): 577-593.Katz MD, Kligman I, Cai L-Q: Paternity by intrauterine insemination with sperm from a man with 5
 -reductase-2 deficiency. The New England Journal of Medicine. 1997, 336 (14): 994-997. 10.1056/NEJM199704033361404.
-reductase-2 deficiency. The New England Journal of Medicine. 1997, 336 (14): 994-997. 10.1056/NEJM199704033361404.Costa EMF, Arnhold IJP, Inácio M, Mendonca BB: Normal bone density in male pseudohermaphroditism due to 5
 -reductase 2 deficiency. Revista do Hospital das Clinicas de Faculdade de Medicina da Universidade de Sao Paulo. 2001, 56 (5): 139-142.
-reductase 2 deficiency. Revista do Hospital das Clinicas de Faculdade de Medicina da Universidade de Sao Paulo. 2001, 56 (5): 139-142.Price P, Wass JAH, Griffin JE: High dose androgen therapy in male pseudohermaphroditism due to 5
 -reductase deficiency and disorders of the androgen receptor. Journal of Clinical Investigation. 1984, 74 (4): 1496-1508. 10.1172/JCI111563.
-reductase deficiency and disorders of the androgen receptor. Journal of Clinical Investigation. 1984, 74 (4): 1496-1508. 10.1172/JCI111563.Cai L-Q, Fratianni CM, Gautier T, Imperato-McGinley J: Dihydrotestosterone regulation of semen in male pseudohermaphrodites with 5
 -reductase-2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1994, 79 (2): 409-414. 10.1210/jc.79.2.409.
-reductase-2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1994, 79 (2): 409-414. 10.1210/jc.79.2.409.Hochberg Z, Chayen R, Reiss N: Clinical, biochemical, and genetic findings in a large pedigree of male and female patients with 5
 -reductase 2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1996, 81 (8): 2821-2827. 10.1210/jc.81.8.2821.
-reductase 2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1996, 81 (8): 2821-2827. 10.1210/jc.81.8.2821.Nordenskjöld A, Ivarsson S-A: Molecular characterization of 5
 -reductase type 2 deficiency and fertility in a Swedish family. The Journal of Clinical Endocrinology & Metabolism. 1998, 83 (9): 3236-3238. 10.1210/jc.83.9.3236.
-reductase type 2 deficiency and fertility in a Swedish family. The Journal of Clinical Endocrinology & Metabolism. 1998, 83 (9): 3236-3238. 10.1210/jc.83.9.3236.Akesode FA, Meyer WJ, Migeon CJ: Male pseudohermaphroditism with gynaecomastia due to testicular 17-ketosteroid reductase deficiency. Clinical Endocrinology. 1977, 7 (6): 443-452. 10.1111/j.1365-2265.1977.tb01336.x.
Imperato-McGinley J, Peterson RE, Stoller R, Goodwin WE: Male pseudohermaphroditism secondary to 17
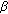 -hydroxysteroid dehydrogenase deficiency: gender role change with puberty. The Journal of Clinical Endocrinology & Metabolism. 1979, 49 (3): 391-395. 10.1210/jcem-49-3-391.
-hydroxysteroid dehydrogenase deficiency: gender role change with puberty. The Journal of Clinical Endocrinology & Metabolism. 1979, 49 (3): 391-395. 10.1210/jcem-49-3-391.Boehmer ALM, Brinkmann AO, Sandkuijl LA: 17
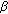 -hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. The Journal of Clinical Endocrinology & Metabolism. 1999, 84 (12): 4713-4721. 10.1210/jc.84.12.4713.
-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. The Journal of Clinical Endocrinology & Metabolism. 1999, 84 (12): 4713-4721. 10.1210/jc.84.12.4713.Cohen-Kettenis PT: Gender change in 46,XY persons with 5
 -reductase-2 deficiency and 17
-reductase-2 deficiency and 17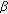 -hydroxysteroid dehydrogenase-3 deficiency. Archives of Sexual Behavior. 2005, 34 (4): 399-410. 10.1007/s10508-005-4339-4.
-hydroxysteroid dehydrogenase-3 deficiency. Archives of Sexual Behavior. 2005, 34 (4): 399-410. 10.1007/s10508-005-4339-4.Mendonca BB, Inácio M, Arnhold IJP: Male pseudohermaphroditism due to 17
 -hydroxysteroid dehydrogenase 3 deficiency: diagnosis, psychological evaluation and management. Medicine. 2000, 79 (5): 299-309. 10.1097/00005792-200009000-00003.
-hydroxysteroid dehydrogenase 3 deficiency: diagnosis, psychological evaluation and management. Medicine. 2000, 79 (5): 299-309. 10.1097/00005792-200009000-00003.Jones GL, Hall JM, Balen AH, Ledger WL: Health-related quality of life measurement in women with polycystic ovary syndrome: a systematic review. Human Reproduction Update. 2008, 14 (1): 15-25. 10.1093/humupd/dmm030.
Nordenskjöld A, Holmdahl G, Frisén L: Type of mutation and surgical procedure affect long-term quality of life for women with congenital adrenal hyperplasia. The Journal of Clinical Endocrinology & Metabolism. 2008, 93 (2): 380-386. 10.1210/jc.2007-0556.
Chachamovich JR, Chachamovich E, Zachia S, Knauth D, Passos EP: What variables predict generic and health-related quality of life in a sample of Brazilian women experiencing infertility?. Human Reproduction. 2007, 22 (7): 1946-1952. 10.1093/humrep/dem080.
Slijper FME, Frets PG, Boehmer ALM, Drop SLS, Niermeijer MF: Androgen insensitivity syndrome (AIS): emotional reactions of parents and adult patients to the clinical diagnosis of AIS and its confirmation by androgen receptor gene mutation analysis. Hormone Research. 2000, 53 (1): 9-15. 10.1159/000023506.
Wagner JL, Chaney JM, Hommel KA: The influence of parental distress on child depressive symptoms in juvenile rheumatic diseases: the moderating effect of illness intrusiveness. Journal of Pediatric Psychology. 2003, 28 (7): 453-462. 10.1093/jpepsy/jsg036.
Thyen U, Lanz K, Holterhus P-M, Hiort O: Epidemiology and initial management of ambiguous genitalia at birth in Germany. Hormone Research. 2006, 66 (4): 195-203. 10.1159/000094782.
 -reductase-2 deficiency syndromes. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (8): 3017-3023. 10.1210/jc.2005-2809.
-reductase-2 deficiency syndromes. The Journal of Clinical Endocrinology & Metabolism. 2006, 91 (8): 3017-3023. 10.1210/jc.2005-2809. -reductase 2 deficiency. Endocrine Reviews. 1993, 14 (5): 577-593.
-reductase 2 deficiency. Endocrine Reviews. 1993, 14 (5): 577-593. -reductase-2 deficiency. The New England Journal of Medicine. 1997, 336 (14): 994-997. 10.1056/NEJM199704033361404.
-reductase-2 deficiency. The New England Journal of Medicine. 1997, 336 (14): 994-997. 10.1056/NEJM199704033361404. -reductase 2 deficiency. Revista do Hospital das Clinicas de Faculdade de Medicina da Universidade de Sao Paulo. 2001, 56 (5): 139-142.
-reductase 2 deficiency. Revista do Hospital das Clinicas de Faculdade de Medicina da Universidade de Sao Paulo. 2001, 56 (5): 139-142. -reductase deficiency and disorders of the androgen receptor. Journal of Clinical Investigation. 1984, 74 (4): 1496-1508. 10.1172/JCI111563.
-reductase deficiency and disorders of the androgen receptor. Journal of Clinical Investigation. 1984, 74 (4): 1496-1508. 10.1172/JCI111563. -reductase-2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1994, 79 (2): 409-414. 10.1210/jc.79.2.409.
-reductase-2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1994, 79 (2): 409-414. 10.1210/jc.79.2.409. -reductase 2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1996, 81 (8): 2821-2827. 10.1210/jc.81.8.2821.
-reductase 2 deficiency. The Journal of Clinical Endocrinology & Metabolism. 1996, 81 (8): 2821-2827. 10.1210/jc.81.8.2821. -reductase type 2 deficiency and fertility in a Swedish family. The Journal of Clinical Endocrinology & Metabolism. 1998, 83 (9): 3236-3238. 10.1210/jc.83.9.3236.
-reductase type 2 deficiency and fertility in a Swedish family. The Journal of Clinical Endocrinology & Metabolism. 1998, 83 (9): 3236-3238. 10.1210/jc.83.9.3236.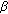 -hydroxysteroid dehydrogenase deficiency: gender role change with puberty. The Journal of Clinical Endocrinology & Metabolism. 1979, 49 (3): 391-395. 10.1210/jcem-49-3-391.
-hydroxysteroid dehydrogenase deficiency: gender role change with puberty. The Journal of Clinical Endocrinology & Metabolism. 1979, 49 (3): 391-395. 10.1210/jcem-49-3-391.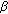 -hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. The Journal of Clinical Endocrinology & Metabolism. 1999, 84 (12): 4713-4721. 10.1210/jc.84.12.4713.
-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. The Journal of Clinical Endocrinology & Metabolism. 1999, 84 (12): 4713-4721. 10.1210/jc.84.12.4713. -reductase-2 deficiency and 17
-reductase-2 deficiency and 17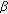 -hydroxysteroid dehydrogenase-3 deficiency. Archives of Sexual Behavior. 2005, 34 (4): 399-410. 10.1007/s10508-005-4339-4.
-hydroxysteroid dehydrogenase-3 deficiency. Archives of Sexual Behavior. 2005, 34 (4): 399-410. 10.1007/s10508-005-4339-4. -hydroxysteroid dehydrogenase 3 deficiency: diagnosis, psychological evaluation and management. Medicine. 2000, 79 (5): 299-309. 10.1097/00005792-200009000-00003.
-hydroxysteroid dehydrogenase 3 deficiency: diagnosis, psychological evaluation and management. Medicine. 2000, 79 (5): 299-309. 10.1097/00005792-200009000-00003.Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wisniewski, A.B., Mazur, T. 46,XY DSD with Female or Ambiguous External Genitalia at Birth due to Androgen Insensitivity Syndrome, 5 -Reductase-2 Deficiency, or 17
-Reductase-2 Deficiency, or 17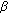 -Hydroxysteroid Dehydrogenase Deficiency: A Review of Quality of Life Outcomes.
Int J Pediatr Endocrinol 2009, 567430 (2009). https://doi.org/10.1155/2009/567430
-Hydroxysteroid Dehydrogenase Deficiency: A Review of Quality of Life Outcomes.
Int J Pediatr Endocrinol 2009, 567430 (2009). https://doi.org/10.1155/2009/567430
Received:
Accepted:
Published:
DOI: https://doi.org/10.1155/2009/567430