- Case Report
- Open access
- Published:
Cushing Syndrome in a 6-Month-Old Infant due to Adrenocortical Tumor
International Journal of Pediatric Endocrinology volume 2009, Article number: 168749 (2009)
Abstract
Cushing syndrome is rare in infancy and usually due to an adrenocortical tumor (ACT). We report an infant with Cushing syndrome due to adrenocortical carcinoma. The patient presented at six months of age with a three-month history of growth failure, rapid weight gain, acne, and irritability. Physical examination showed obesity, hypertension, and Cushingoid features. Biochemical evaluation showed very high serum cortisol, mildly elevated testosterone, and suppressed ACTH. Abdominal MRI revealed a heterogeneous right adrenal mass extending into the inferior vena cava. Evaluation for metastases was negative. The tumor was removed surgically en bloc. Pathologic examination demonstrated low mitotic rate, but capsular and vascular invasion. She received no adjuvant therapy. Her linear growth has improved and Cushingoid features resolved. Hormonal markers and quarterly PET scans have been negative for recurrence 24 months postoperatively. In conclusion, adrenocortical neoplasms in children are rare, but should be considered in the differential diagnosis of Cushing syndrome.
1. Introduction
Adrenocortical carcinoma is a rare malignancy comprising only about 0.2% of all cases of childhood cancer. This rare malignancy is associated with a high rate of recurrence and mortality with a reported 5-year event-free survival estimate of 54.2% in children. Pediatric adrenocortical tumors (ACTs) occur most commonly in females and in children less than four years [1]. Eighty to ninety percent of patients have functional tumors with endocrine manifestations at diagnosis and the majority present with virilization, alone or in combination with overproduction of other adrenal hormones. Isolated Cushing syndrome is rare [1, 2]. We report an infant with Cushing syndrome without the clinical features of androgen or mineralocorticoid excess, consequently diagnosed with adrenocortical carcinoma.
2. Case Report
The patient is a Caucasian female born at term with a birth weight of 3.06 kg; pregnancy and delivery were uncomplicated. At six months of age, she was referred to pediatric endocrine clinic due to features of Cushing syndrome. She had a three month history of progressive Cushingoid facies, acne and irritability. Her linear growth apparently had been arrested between three and six months of age, although she gained weight at an accelerated rate (Figure 1). Prior to referral, laboratory evaluation by her primary care physician revealed serum cortisol of 240 mcg/dl (normal, 4.5–22.7 mcg/dl); total testosterone of 185 ng/dl (normal, 6–77ng/dl), and DHEA-S of 401 mcg/dl (normal, 16–96 mcg/dl). Serum ACTH was less than 5 pg/ml (normal, 10–60 pg/ml).

Growth chart demonstrating impairment of linear growth, a classic feature of Cushing Syndrome, prior to diagnosis. After surgical removal of tumor, the patient exhibited catch-up growth during the following six months.
Her weight at initial evaluation in our clinic at six months of age was 7.52 kg (60–75th percentile) and length was 59.1 cm ( 5th percentile). Blood pressure was 110/70 mmHg. Physical examination showed Cushingoid facies, buffalo hump, facial acne, and poor muscle tone (Figure 2). There were no signs of virilization. Family history was noncontributory.
5th percentile). Blood pressure was 110/70 mmHg. Physical examination showed Cushingoid facies, buffalo hump, facial acne, and poor muscle tone (Figure 2). There were no signs of virilization. Family history was noncontributory.

Physical examination of the patient revealed features of Cushing Syndrome including round facies, acne, plethora, central obesity, and poor muscle tone; she lacked clitoromegaly, hirsutism or other signs of virilization.
An MRI of the abdomen showed a heterogeneous right adrenal mass measuring  this mass was found to extend as a tumor thrombus into the inferior vena cava. The tumor thrombus appeared to extend to approximately 2 cm from the right atrium. There were no metastases seen in the liver or lungs on MRI. A Doppler ultrasound showed minimal, but present flow around the IVC tumor thrombus which measured
this mass was found to extend as a tumor thrombus into the inferior vena cava. The tumor thrombus appeared to extend to approximately 2 cm from the right atrium. There were no metastases seen in the liver or lungs on MRI. A Doppler ultrasound showed minimal, but present flow around the IVC tumor thrombus which measured  Laboratory evaluation showed serum cortisol 70.8 mcg/dl (normal, 4.5–22.7 mcg/dl); aldosterone 4.2 ng/dl (normal 6.5–86.0 ng/dl); testosterone 59 ng/dl (normal, 6–77 ng/dl); ACTH
Laboratory evaluation showed serum cortisol 70.8 mcg/dl (normal, 4.5–22.7 mcg/dl); aldosterone 4.2 ng/dl (normal 6.5–86.0 ng/dl); testosterone 59 ng/dl (normal, 6–77 ng/dl); ACTH  (normal, 10–60 pg/ml).
(normal, 10–60 pg/ml).
The patient underwent surgical resection of the mass ten days after diagnosis. She was prescribed ketoconozole prior to surgery due to moderately elevated blood pressure. However, this was not well tolerated by the patient and was discontinued.
Surgical resection was undertaken and intraoperative findings showed a large, well-circumscribed mass in the right adrenal gland measuring  mass which did not invade the liver, kidney or other surrounding soft tissues. On intraoperative transesophageal echocardiography, there appeared to be thickening of the wall of the superior vena cava of unknown significance. The intracaval clot did not extend to the right atrium and cardiopulmonary bypass was not required. Blood flow was visualized around the caval clot with no evidence of adherence to the caval wall. The adrenal mass and thrombus were removed en bloc (Figure 3(a)).
mass which did not invade the liver, kidney or other surrounding soft tissues. On intraoperative transesophageal echocardiography, there appeared to be thickening of the wall of the superior vena cava of unknown significance. The intracaval clot did not extend to the right atrium and cardiopulmonary bypass was not required. Blood flow was visualized around the caval clot with no evidence of adherence to the caval wall. The adrenal mass and thrombus were removed en bloc (Figure 3(a)).

(a) Grossly, the resected specimen showed a well-circumscribed tumor without normal adrenal gland. The tumor measured 5.2×4×3.5 cm and weighed 43 gm. (b) Adrenal cortical carcinoma with necrosis (upper field) and coarse calcifications. (c) Adrenal cortical carcinoma. Richly vascular neoplasm composed of nodules of tumor cells with lightly eosinophilic cytoplasm. The tumor contains bands of fibrous tissue and coarse calcifications (Hematoxylin and eosin, 20X).
Pathologic examination revealed a 43 gram mass which demonstrated capsular and vascular invasion. The mass showed five to ten percent necrosis of the mass with spotty calcifications (Figures 3(b) and 3(c)). The surgical margins were negative. She received stress dosing with hydrocortisone during and after surgery; she was reduced to a physiologic replacement dose by 15 days postoperatively.
She was considered to have Stage 1 adrenocortical carcinoma based on the relatively small tumor size, complete surgical resection, lack of metastases, and normalization of hormonal markers postoperatively. The tumor cells demonstrated a low mitotic count ( to 2 per 50 high power fields and no atypical mitoses). In addition, the MIB-1 index was low at
to 2 per 50 high power fields and no atypical mitoses). In addition, the MIB-1 index was low at  to 12% with regional variability. Tumor immunohistochemistry for p53 protein showed weak staining with no accumulation in the nucleus; when p53 amplification is present, the defective proteins accumulate within the nucleus due to abnormal cellular processing. To evaluate for a germline p53 mutation in the patient, whole gene sequencing was performed for the TP53 gene and did not reveal any known mutation. No adjunctive therapies were recommended. Quarterly PET scanning to monitor for recurrence is underway, and has been negative for 24 months post-operatively.
to 12% with regional variability. Tumor immunohistochemistry for p53 protein showed weak staining with no accumulation in the nucleus; when p53 amplification is present, the defective proteins accumulate within the nucleus due to abnormal cellular processing. To evaluate for a germline p53 mutation in the patient, whole gene sequencing was performed for the TP53 gene and did not reveal any known mutation. No adjunctive therapies were recommended. Quarterly PET scanning to monitor for recurrence is underway, and has been negative for 24 months post-operatively.
Adrenal markers were measured after surgery and at monthly intervals. Two months after surgery, laboratory evaluation showed total testosterone level of  ng/dl (normal, 6–77 ng/dl); DHEA <1.0ng/ml (normal,
ng/dl (normal, 6–77 ng/dl); DHEA <1.0ng/ml (normal,  ng/ml); DHEA-S <15.0 ng/dl (normal, 16–96 mcg/dl). ACTH was 7.6 pg/ml (normal, 10–60 pg/ml). Hormonal markers remain normal up until time of publication. Growth velocity began to increase within two months after surgery, and five months after surgery was 25 cm/year consistent with catch-up growth.
ng/ml); DHEA-S <15.0 ng/dl (normal, 16–96 mcg/dl). ACTH was 7.6 pg/ml (normal, 10–60 pg/ml). Hormonal markers remain normal up until time of publication. Growth velocity began to increase within two months after surgery, and five months after surgery was 25 cm/year consistent with catch-up growth.
3. Discussion
Our patient demonstrated classic features of glucocorticoid excess including growth failure, hypertension, acne, plethora and a buffalo hump with excessive weight gain. She had biochemical evidence for excessive glucocorticoid production. The differential diagnosis of Cushing syndrome in an infant includes adrenocortical tumors (carcinoma or adenomas), as well as multinodular adrenal hyperplasia. In addition, Cushing disease, ectopic ACTH production, and iatrogenic Cushing syndrome should be considered. Miller et al., demonstrated that of 60 infants with Cushing syndrome, 48 had adrenal tumors. Two had ectopic ACTH production, five had nodular adrenal hyperplasia, four had undefined adrenal hyperplasia and one had an ACTH producing tumor [3]. Therefore, first an adrenocortical tumor should be excluded in an infant with Cushing syndrome as long as there is no history for exposure to exogenous glucocorticoids. In our case, suppressed ACTH level was consistent with a peripheral source. The unilateral adrenal mass in the MRI indicated an adrenal tumor and histopathological findings confirmed an adrenocortical carcinoma.
Pediatric adrenocortical tumors have distinct clinical and biological features in comparison to those which occur in adults. It is estimated that there are 19–20 new cases of adrenocortical carcinoma in children and adolescents per year in the United States according to the Surveillance, Epidemiology and End Results Program. Most of these patients are either toddlers, early school age children or in their late teens as pediatric adrenocortical tumors have a bimodal age distribution. There is a peak in incidence before four years of age (0.4 cases per million) which then declines during the subsequent ten years. The incidence then rises to 0.2 per million during the late teens. Interestingly, the majority (60%) of pediatric adrenocortical tumors occur in children less than five years which is similar to the distribution seen with tumors of embryonic origin [1].
Although our patient fits the common age distribution for an adrenocortical tumor, her presentation with Cushingoid features alone is unusual especially at such a young age. Although biochemically she demonstrated elevations in both androgens and cortisol, she had no evidence of virilization on physical examination. Michalkiewicz et al., found in a registry of 254 pediatric patients with ACTs that 55% presented with virilization alone. Twenty-nine percent presented with mixed overproduction of adrenal hormones. Only 5.5% percent of this group presented with isolated Cushing syndrome, and this tended to occur in older children (median age, 12.6 years). In addition, Lefevre et al., showed similar results in their analysis of 42 children with ACTs in France. More than 90% of patients presented with virilization. Therefore, our patient is remarkable in that she presented with Cushingoid features alone during infancy [1].
The tendency for adrenocortical tumors to occur in younger age groups and to be predominantly androgen-producing could be linked to their relationship to the fetal adrenal cortex. The fetal adrenal cortex is composed of two distinct zones: the outer definitive zone which is steroidogenically quiescent until late gestation and an inner zone which appears to produce steroid hormones throughout gestation. The inner fetal zone makes up 85–90% of the total fetal adrenal cortex at birth from which time it begins to undergo apoptosis. The histopathologic features of ACTs in young children suggest that they arise from the fetal zone which has a tendency towards androgen production due to differential expression of steroidogenic enzymes at different times in development. The primary steroid product of the fetal adrenal cortex is dehydroepiandrosterone sulfate (DHEA-S) which is the precursor of placental estrogen [4]. Thus, the derivation of adrenocortical tumors of infants, and young children from the fetal zone of the adrenal cortex could help to explain their preferential elaboration of androgens.
Our patient's tumor was negative for p53 mutation which is estimated to occur in 80–90% of all pediatric adrenocortical tumors. Interestingly, a distinct germline p53 mutation, TP53 R337H, has been described in Brazilian patients with adrenocortical tumors [5, 6]. This mutation leads to an abnormal folding of the TP53 protein and accumulation in the nucleus; loss of heterozygosity at this locus is though to be involved in tumorigenesis. In a study evaluating penetrance of this mutation, 10% of 240 carriers of this mutation developed adrenocortical tumors [7].
Our patient has a favorable prognosis given complete surgical resection, small tumor size, lack of metastasis and young age. From the International Pediatric Adrenocortical Tumor Registry report including data from 254 patients, those with completely resected tumors weighing less than 200 gm and without metastasis, had a five year event free survival rate of 91%. This study also showed that age less than four years was independently associated with better prognosis; multivariate analysis showed adjusted odds ratio of 2.6 for age less than four years [1]. Therefore, our patient has an excellent prognosis. As mentioned earlier, her follow-up PET scans and hormonal markers have remained negative for 24 months postoperatively.
In conclusion, although rare, ACTs should be considered in the differential diagnosis of Cushing syndrome in the pediatric age group. While virilizing features are typically dominant, ACTs may present with isolated Cushing syndrome. Early diagnosis, adequate perioperative management with specific emphasis on postoperative glucocorticoid replacement, complete excision of tumor and close follow-up for recurrence and metastases are crucial to improve survival.
References
Michalkiewicz E, Sandrini R, Figueiredo B: Clinical and outcome characteristics of children with adrenocortical tumors: a report from the international pediatric adrenocortical tumor registry. Journal of Clinical Oncology. 2004, 22 (5): 838-845. 10.1200/JCO.2004.08.085.
Teinturier C, Pauchard MS, Brugières L, Landais P, Chaussain JL, Bougnères PF: Clinical and prognostic aspects of adrenocortical neoplasms in childhood. Medical and Pediatric Oncology. 1999, 32 (2): 106-111.
Miller WL, Townsend JJ, Grumbach MM, Kaplan SL: An infant with Cushing's disease due to an adrenocorticotropin-producing pituitary adenoma. Journal of Clinical Endocrinology and Metabolism. 1979, 48 (6): 1017-1025. 10.1210/jcem-48-6-1017.
Coulter CL: Fetal adrenal development: insight gained from adrenal tumors. Trends in Endocrinology and Metabolism. 2005, 16 (5): 235-242. 10.1016/j.tem.2005.05.010.
Ribeiro RC, Sandrini F, Figueiredo B: An inherited
 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2001, 98 (16): 9330-9335. 10.1073/pnas.161479898.
mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2001, 98 (16): 9330-9335. 10.1073/pnas.161479898.DiGiammarino EL, Lee AS, Cadwell C: A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nature Structural Biology. 2002, 9 (1): 12-16. 10.1038/nsb730.
Figueiredo BC, Sandrini R, Zambetti GP: Penetrance of adrenocortical tumours associated with the germline
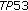 R337H mutation. Journal of Medical Genetics. 2006, 43 (1): 91-96.
R337H mutation. Journal of Medical Genetics. 2006, 43 (1): 91-96.
 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2001, 98 (16): 9330-9335. 10.1073/pnas.161479898.
mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2001, 98 (16): 9330-9335. 10.1073/pnas.161479898.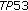 R337H mutation. Journal of Medical Genetics. 2006, 43 (1): 91-96.
R337H mutation. Journal of Medical Genetics. 2006, 43 (1): 91-96.Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fudge, E., von Allmen, D., Volmar, K. et al. Cushing Syndrome in a 6-Month-Old Infant due to Adrenocortical Tumor. Int J Pediatr Endocrinol 2009, 168749 (2009). https://doi.org/10.1155/2009/168749
Received:
Accepted:
Published:
DOI: https://doi.org/10.1155/2009/168749